In RNA-Seq experiments, the ultimate goal is to accurately quantify the abundance of RNA transcripts in each sample. During the library generation process, PCR is used to amplify, or copy, transcripts so they can be abundant enough for both quality control and sequencing. During the amplification process, copies are made from identical fragments of the original molecule. As these copies are indistinguishable, it is extremely challenging to determine the original number of molecules in the sample. The use of Unique Molecular Identifiers (UMIs) is an established solution to quantify these original molecules, especially in low-input experiments such as single-cell RNA-Seq.
1. What are UMIs?
UMIs, also known as Molecular Barcodes or Random Barcodes, consist of short random nucleotide sequences which are added to each molecule in a sample as a unique tag. The UMIs are introduced during library generation before the final library fragment is amplified in the PCR step (Fig. 1). This idea was first implemented in an iCLIP protocol1, a cross-linking and immuno-precipitation method for studying specific protein-RNA interactions.

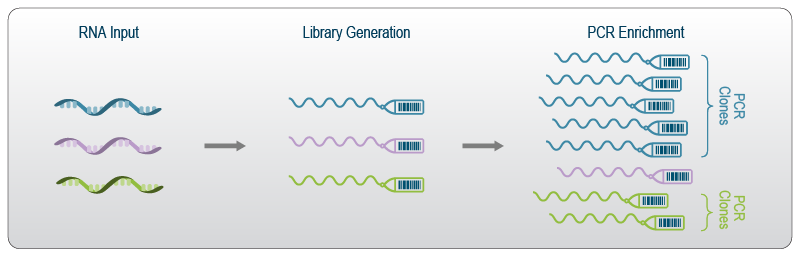
Figure 1 | Individual RNA molecules in each sample are tagged with a unique barcode. These barcodes are copied along with the molecule in the PCR step. Downstream data analysis can then deduplicate the copies, revealing the original ratio of molecules in the sample and eliminating amplification bias.
The primary advantage of including UMIs in a sequencing experiment is to enable the accurate bioinformatic identification of PCR duplicates. Without this capacity, the PCR duplicates can have a detrimental impact on downstream data analysis, especially when amplification biases occurred. Biases in the PCR reaction step can lead to overrepresentation of particular sequences in the final library2 due to preferential over-amplification. To prevent this bias from further propagation, it has been proposed to remove reads or read pairs with the exact same alignment coordinate, as they are predicted to arise through the PCR amplification of the same molecule3. During the later cycles of PCR, error rates increase, and biases can manifest even further (check out the PCR section in LEXICON Chapter 7 for more details).
UMIs therefore ultimately act as tags that allow the accurate identification following the removal of PCR duplicates in sequencing data. Thereby, the data quality can be improved by revealing the original number and ratio of molecules in the sample. Despite the ability to rescue some effects of excessive amplification by removing duplicates, biases in the data can be minimized by using the correct PCR cycle number. To learn more about how the optimal cycle number for PCR is determined, visit our blog article.
2. Why are UMIs Useful? Some Applications for UMI in RNA-Seq
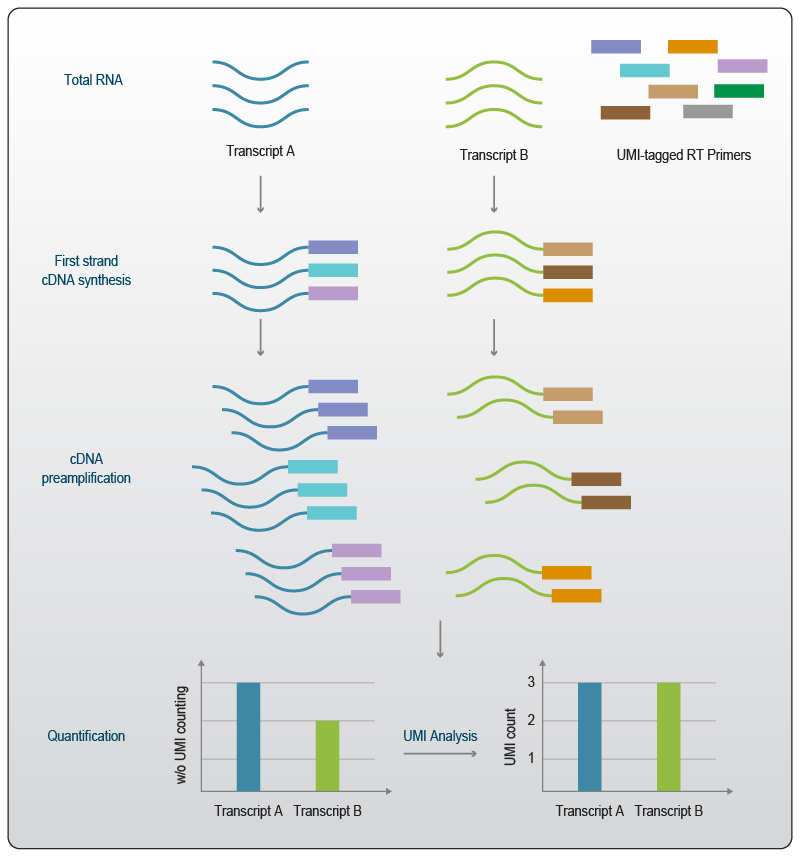
UMIs enable the quantification of the absolute number of molecules in a sample without the need to detect each individual molecule or identify the number of copies made from them. While measuring the number of copies of each sequence is challenging, counting the number of distinct UMI sequences is easier, and this information does not get lost during the amplification process (Fig. 2). Further, normalization of such RNA-Seq datasets can be performed without the loss of accuracy4.
Figure 2 | Transcript level quantification with UMIs. Transcripts or cDNAs are tagged with UMIs in an early step of library generation. The UMI sequences can then be used for quantification of the number of molecules that were originally present in the sample. UMIs can thus control for amplification biases associated with PCR-based sample preparation. Adapted from 5.
Analyzing UMIs is a convenient method of detection and measurement of the abundance of individual molecules present in a complex sample mixture even without an amplification step. Further, different RNA species are present in the sample at different concentrations. It was estimated that differences in concentration between high and low abundant mRNAs in a cell or tissue may vary within 6 to 10 orders of magnitude. These differences in concentrations make the molecular counting procedure via sequencing difficult4 and can benefit from the use of UMIs. Thus, UMIs may be utilized in any sequencing method, where confident identification of duplicates by alignment coordinate is not possible or where accurate quantification is required. The UMI method could be applied to count all types of molecules or particles such as viruses, proteins, and in methods like ChiP-Seq, karyotyping and others4.
Another application benefiting from using UMIs is targeted RNA-Seq, i.e., when libraries are prepared from more restricted regions of the transcript. In these cases, there is a higher chance that identical priming occurs on unique transcripts or first strand cDNA molecules than when using protocols for whole transcriptome RNA-Seq. This results in sequencing reads with identical mapping coordinates and sequences. Without UMIs, these reads may not be quantified correctly, resulting in inaccurate read count data. Including UMIs during library generation, however, clearly distinguishes unique priming events from PCR duplicates and allows for accurate quantification of sequencing reads.
Targeted sequencing can also be used to assess rare variants carrying mutations that can cause a variety of diseases, and are of particular importance in cancer and oncology. Due to the fact that several steps of the RNA-sequencing workflow can introduce errors, the identification of true rare mutations present in the original RNA molecule can be quite challenging. UMIs are particularly useful to discriminate between errors introduced by the workflow and mutations present in the original molecule6. Variants or mutations are considered “true” when they are identical within the individual reads carrying the same UMI and between reads with different UMIs (Fig. 3). Finding the same mutation in reads with different UMIs can further be used to exclude systematic errors introduced by reverse transcription in the first strand synthesis step.
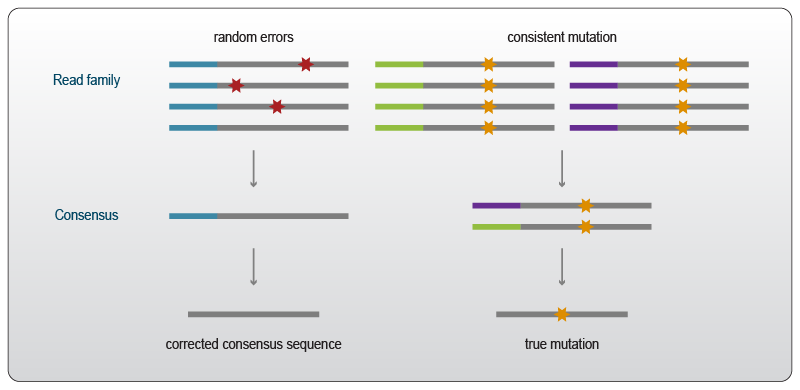
Figure 3 | Alignment of read families sorted by UMIs (color-coded) enables the discrimination of rare variants from random errors introduced during the library preparation and sequencing workflow. True mutations occur throughout the reads carrying the same UMI and can be seen also in reads containing a different UMI.
(Adapted from 6).
3. UMIs in Single-cell RNA Sequencing
UMIs were shown to reduce the amplification noise in single-cell RNA-Seq studies7 and here, UMIs are particularly useful. The amplification bias is one of the main challenges for single-cell experiments and many protocols use multiple, consecutive PCR amplification steps.
A typical single mammalian cell contains approximately 105 – 106 mRNA molecules. The Human Cell Atlas determined ~11,000 detectable genes in various human cell lines. As genes can be expressed by multiple transcript isoforms differing in their transcriptional start and end sites, exon / intron composition, and expression level, the quantification of transcripts in single cells is particularly challenging. For single-cell experiments, the quantification of genes is often used as more accurate and easier. In any case, to estimate the number of genes or transcripts expressed in a single cell, UMIs are crucial.
4. How Many Different UMIs are Needed?
UMIs will reflect molecule counts only if the number of available distinct tags is substantially larger than the typical number of identical molecules. The random sequence composition of the UMIs ensures that every library fragment-UMI combination is unique. For this, a large number of random UMI sequences needs to be available.
As an example, for UMI sequences of a length of 10 random nucleotides, as present in QuantSeq-Pool, 410 or 1,048,576 different UMI sequences are used.
It is important to note that the incorporation of the UMI into any library preparation does not interfere with the RNA-seq process, and similar counts of reads mapping to each gene were seen in both UMI-tagged and untagged samples4. Since UMIs are agnostic to the library generation chemistry, they are compatible with any indexing strategy, using single or dual indexing and / or additional sample-barcodes (also termed inline indices or sample indices).
5. UMIs and the Absolute Truth – Implications for Data Analysis
A fundamental assumption in RNA-Seq has been that library fragments sharing a UMI sequence and read mapping locus were derived from the same initial input molecule. However, it is now clear that a fraction of sequencing reads sharing the same unique molecular identifier would map to different, but closely spaced locations. Due to errors occurring during the sequencing process, the mapping coordinates are not always precise (see Chapter 2 – Next Generation Sequencing for more details).
This imprecision may be misleading for the commonly used analysis tools, which eliminate PCR duplicates and perform counting under the assumption that reads with different mapping coordinates are derived from different starting molecules. This could result in overestimating the expression levels of low abundant transcripts by a large factor and highlights the need for an accurate data analysis pipeline for UMIs in RNA-Seq projects8.
Some UMI data analysis tools also utilize error correcting functions to account for errors that alter the UMI sequence. For accurate quantification it is beneficial to trace back the erroneous UMI to its parent sequence (Fig. 4).
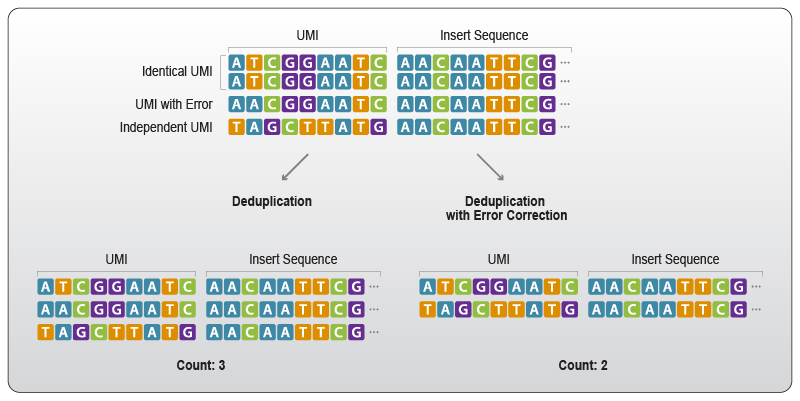
Figure 4 | Errors within the UMI sequence can be corrected for more accurate quantification. Erroneous UMIs can be identified and reverted to the original sequence prior to deduplication. This is done by assessing the distance between the individual sequences and the proposed parental sequence. Deduplication is then based on the parental UMI sequence and also removes reads with slight alterations in the UMI sequence (e.g., caused by nucleotide mis-incorporation).
The knowledge that read mapping coordinates may shift by few bases can also be integrated into the analysis.
6. Some Actionable Advice: When are UMIs Useful for RNA-Seq Library Preps?
UMIs are mostly used to remove PCR duplicates with the aim to reduce amplification bias and to estimate how many genes / transcripts are expressed in single cells. Therefore, UMIs are most useful when the input amounts are limiting (single-cell level to low input amounts of ≤ 10 ng total RNA), while UMIs may not offer a clear benefit for higher input amounts as the number of RNA molecules in this case exceeds the number of possible UMI sequences. UMIs can also indicate over-sequencing, i.e., when the sequencing depth is too high in comparison to the library complexity. While over-sequencing is not harmful per se, avoiding over-sequencing reduces costs and frees up sequencing space that can be used to include more replicates to increase the statistical power. UMIs can also help to estimate accessible transcripts, e.g., RNA derived from FFPE samples is very heterogeneous and the number of accessible transcripts varies from sample to sample due to cross-linking.
Library preparation methods that contain built-in UMIs are versatile and can be used for all samples. Processing and deduplicating UMIs is usually optional and can be omitted when working with high input amounts to save computational resources.

Literature:
1 König, J., Zarnack, K., Rot, G., et al., (2010) iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 17:909-915. doi: 10.1038/nsmb.1838. Epub. PMID: 20601959; PMCID: PMC3000544.
2 Aird, D., Ross, M.G., Chen, WS. et al. (2011) Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol 12, R18. doi: 10.1186/gb-2011-12-2-r18.
3 Sims, D., Sudbery, I., Ilott, N.E., et al., (2014). Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 15:121-132. doi: 10.1038/nrg3642. PMID: 24434847.
4 Kivioja, T., Vähärautio, A., Karlsson, K. et al. (2012). Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 9, 72–74, doi: 10.1038/nmeth.1778.
5 Kolodziejczyk, A.A., and Lönnberg, T. (2018) Global and targeted approaches to single-cell transcriptome characterization, Briefings in Functional Genomics,17: 209- 219, doi: 10.1093/bfgp/elx025.
6 Roloff, G. W., Lai, C., Hourigan, C. S., et al. (2017) Technical advances in the measurement of residual disease in acute myeloid leukemia. Journal of clinical medicine, 6: 87. doi:10.3390/jcm6090087.
7 Islam, S., Zeisel, A., Joost, S. et al. (2014) Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods 11, 163–166, doi: 10.1038/nmeth.2772.
8 Sena, J.A., Galotto, G., Devitt, N.P. et al. (2018) Unique Molecular Identifiers reveal a novel sequencing artefact with implications for RNA-Seq based gene expression analysis. Sci Rep 8, 13121, doi: 10.1038/s41598-018-31064-7.
human cell atlas: https://www.proteinatlas.org/humanproteome/cell/cell+line
Written by Dr. Yvonne Goepel and Alexander Mojcher
Are you ready to become an RNA Expert?
Sign up and gain access to helpful checklists in PDF format that can assist you in your experiments. In addition, you’ll have the opportunity to download the RNA LEXICON E-BOOK in PDF format as well.




