While protocols to generate Next Generation Sequencing (NGS) libraries for RNA sequencing (RNA-Seq) have been highly optimized over the past decade, the conversion of RNA to cDNA remains a limiting step. In most RNA-Seq workflows, a combination of restricted RNA input and low reverse transcription efficiency calls for PCR amplification to produce sufficient library to start the NGS analysis.
The required number of PCR cycles depends mostly on the amount of initial cDNA template, which in turn is proportional to the initial RNA input in optimized, reproducible RNA-Seq library preparation workflows. However, other sample properties can also affect cDNA synthesis such as:
- Integrity (e.g., degradation & fragmentation),
- Target sequence content (e.g., fraction of polyadenylated RNAs),
- Purity (e. g., occurrence of PCR inhibitors), and
- Sequence accessibility (e.g., crosslinked nucleotides in FFPE-derived samples).
Further, automation of the RNA-Seq library preparation process on robotic liquid handling platforms is known to reduce the overall library preparation efficiency. This results in the need to increase the number of PCR amplification rounds by 1-2 cycles.
In many cases, adaption of the PCR cycle number is necessary to account for the varying content of amplifiable target sequences in the samples. Exceptions are homogeneous RNA-Seq experiments such as screening setups with RNA of comparable equality extracted from a similar number of cells. Here, a correct PCR cycle number determined in an initial assessment or derived from literature can be used.
Determining the correct PCR cycle number
PCR amplification is commonly used to generate NGS-ready libraries by introducing sequences required for cluster formation and indices for multiplexing. To avoid PCR duplicates and artefacts care must be taken to amplify using the correct number of PCR cycles. This number is best determined by a qPCR assay (Fig. 1).

Figure 1 | Determining the correct number of cycles for end-point PCR. By using e.g., 1.7 μl of library cDNA for qPCR, the cycle number corresponding to 50 % of the maximum fluorescence can be determined (15 cycles). In end-point PCR, the remaining 17 μl of purified cDNA template should therefore be amplified with 12 cycles (15 – 3 cycles) to account for the difference in template concentration. Above data reproduced from CORALL Total RNA-Seq Library Prep Kit User Guide (095UG190V0130).
Using the wrong number of PCR cycles for RNA-Seq library amplification
Undercycling can lead to low yield, and although re-amplification is possible to “rescue” the library generation, this leads to an unnecessary high number of overall PCR cycles.
Overcycling occurs when PCR primers become exhausted but cycling continues.
- In case dNTPs are still present, the reaction proceeds with PCR products priming themselves, creating longer PCR artifacts with chimeric sequences (Kanagawa 2003).
In case the dNTPs concentration becomes limiting as well, “bubble products” can appear in over-cycled PCR reactions, indicating the presence of heteroduplexes composed of only partially homologous library fragments. Without the competition of PCR primers, single-stranded PCR products carrying complementary adapter sequences at their 5’ and 3’ ends will anneal to corresponding template, creating bubble-like conformations. See the FAQ section of the UC Davis Genome Center for an example.
Detecting PCR overcycling
Library overcycling can be determined in gel separation- or microfluidics-based analyses. When running in its native state, an accurately cycled library should show only one peak corresponding to the desired library length. Overcycling is indicated by either a smear of longer or shorter products (caused by product-priming) or by the occurrence of a distinct, second peak migrating slower than the peak with the desired products (Fig 2. and Fig. 3).

Figure 2 | Bioanalyzer traces of correctly amplified and over-cycled 3’ mRNA-Seq libraries. QuantSeq 3’ mRNA-Seq libraries were amplified from templates created with 10 ng UHRR input with either the correct number of PCR cycles (19 cycles, blue trace) or over-cycled (24 cycles, red trace). Note the high molecular weight smear extending beyond the upper marker.

Figure 3 | Bioanalyzer traces of over-cycled mRNA-Seq libraries. Bioanalyzer traces of SENSE mRNA-Seq for Illumina libraries with a second peak in high molecular weight regions due to overcycling. Libraries amplified from cDNA templates synthesized with RTS (red) or RTL (blue) buffer.
Rescuing over-cycled RNA-Seq libraries
If over-cycled RNA-Seq libraries show a distinct second peak corresponding to “bubble products”, a “reconditioning” PCR with one or very few PCR cycles can be performed to yield perfectly double-stranded PCR products (Thompson et al., 2002). If the over-cycled library contains PCR products that stem from PCR product priming (due to primer depletion), a rescue of this fraction is not possible.
Effects of library PCR overcycling
Library quantification – Over-cycled PCR libraries are difficult to quantify accurately using standard microfluidics or gel-based methods, since the products are heterogeneous in structure and migration. qPCR assays are the method of choice, if over-cycled libraries have to be sequenced, since they will measure only the amplifiable fraction, irrespective of product structures. However, if the over-cycled library contains PCR products primed by other PCR products, then these chimeric results, while counted in the qPCR assay, can be too long to cluster on a short-read platform. Sequencing is still possible, but the read share can be expected to not match the calculation.
Mapping and gene expression quantification – Reads from chimeric PCR products might be mapped incorrectly or not at all, affecting the accuracy of gene expression quantification and leading to incorrect biological conclusions (Fig. 4). External RNA-Seq controls such as Lexogen’s SIRV spike-in transcripts can help to detect and quantify these effects.
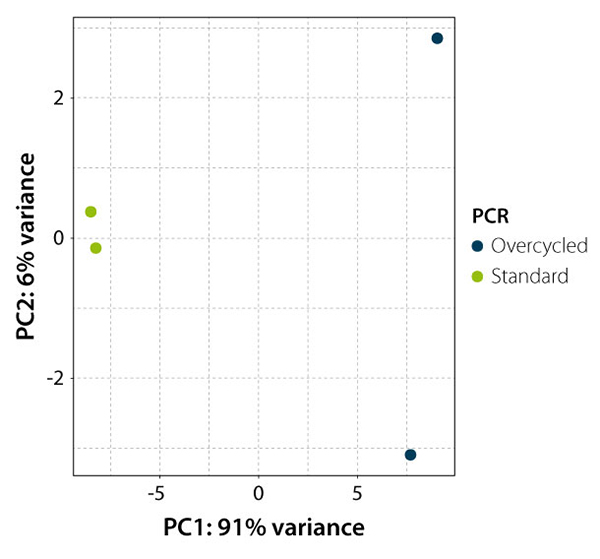
Figure 4 | Principal Component Analysis of standard and over-cycled RNA-Seq libraries. QuantSeq 3’ mRNA-Seq libraries produced from the same input were either amplified correctly (green dots) or over-cycled (dark blue). Gene expression values were then evaluated in a Principal Component Analysis (PCA). The Principal Component 1 (PC1) on the x-axis clearly separates both conditions, explaining 91 % of the variability seen in the read counts of the standard and the over-cycled samples. PC2 shows that the two over-cycled libraries do not cluster (unlike the correctly cycled libraries). This indicates that overcycling caused differential bias between the two incorrectly amplified samples, most likely by a variation in the duplicates fraction.
Summary
Correct PCR amplification is not only necessary to maintain the initial complexity of RNA-Seq libraries and to keep the number of PCR duplicates low, but to avoid the production of artefacts due to the depletion of PCR primers and/or dNTPs. These artefacts can affect library quantification, lane-mixing, generation of clusters on short-read NGS platforms, mapping, and eventually gene expression detection and quantification. qPCR is the best method to determine the correct PCR cycle number to avoid overcycling and under-cycling, and once this number is established, it can be used for RNA samples similar in amount, target sequence fraction, and integrity.
Learn more about Lexogen RNA-Seq Library Prep Kits and PCR Add-on Kit:
- CORALL Total RNA-Seq Library Prep Kit
- QuantSeq 3′ mRNA-Seq Library Prep Kit FWD for Illumina
- QuantSeq 3′ mRNA-Seq Library Prep Kit REV for Illumina
- QuantSeq-Flex Targeted RNA-Seq Library Prep Kit V2 for Illumina
- QuantSeq-Pool Sample-Barcoded 3′ mRNA-Seq Library Prep Kit for Illumina
- PCR Add-on Kit for Illumina
- Lexogen 12 nt Unique Dual Index System (UDI) for RNA-Seq
References:






