SIRVs
The most comprehensive
RNA spike-in controls

SIRVs Introduction
Biases in RNA Sequencing
Spike-in Transcripts in RNA-Seq
SIRV Modules: Isoforms, ERCCs, and long SIRVs Modules
Transcriptomes are complex and consist of several RNA classes with specific properties. Spike-in RNA controls must reflect these to be representative for a given experimental design. The Spike-In RNA Variants (SIRV) were conceived as a family of modules to offer tailored solutions for the control of RNA-seq experiments. SIRVs are available as an isoform module, which contains a group of synthetic transcripts that mimic transcriptome complexity, and as a length module to cover transcript lengths of up to 12 kb. While the SIRV isoform module is available as a stand-alone module (Cat. No. 050) or mixed with ERCCs to additionally mimic abundance complexity (Cat. No. 051), the long SIRVs module is provided in a mix together with the SIRV isoform module and the ERCC module (Cat. No. 141). See Modular Design for more information on the spike-in concept and SIRV Sets for details on the mixes available to users.
Spike-in Experiment Rationales
Here, we describe considerations for planning RNA-Seq experiments. However, the SIRV mixes are not only suitable for assessing NGS setups but also for quantification on microarray platforms and in qPCR assays.
Spiking of samples
SIRVs are spiked into samples before library preparation, either to purified RNA or at an upstream processing stage such as homogenization (e.g. RNA extraction from tissues or fluids) or lysis (e.g. single cell applications). Due to their sequences being non-identical to genomic and transcriptomic database entries they can be combined with RNA from any organism (see Modular Design for details). Since the SIRV RNAs are polyadenylated, library preparation can start from poly(A)-selected fractions as well as from total RNA, depleted RNA, etc.
Typically, the amount of spike-in RNA is adjusted to have only 1 % of all NGS reads mapping to the SIRV genome, the “SIRVome”. This might be increased to 2-5% for setups with low read depth (< 5M reads) that analyze SIRV-Set 3 or SIRV-Set 4, which contain more than one SIRV module. The spike-in amounts are best tailored to the RNA fractions of interest (e.g. total RNA, ribosomal depleted RNA or poly(A)-enriched RNA) and to the amount of sample. Alternatively, spike-in amounts might be kept constant to measure variations in the sample like the mRNA content or metabolic states.
Library Preparation and Sequencing
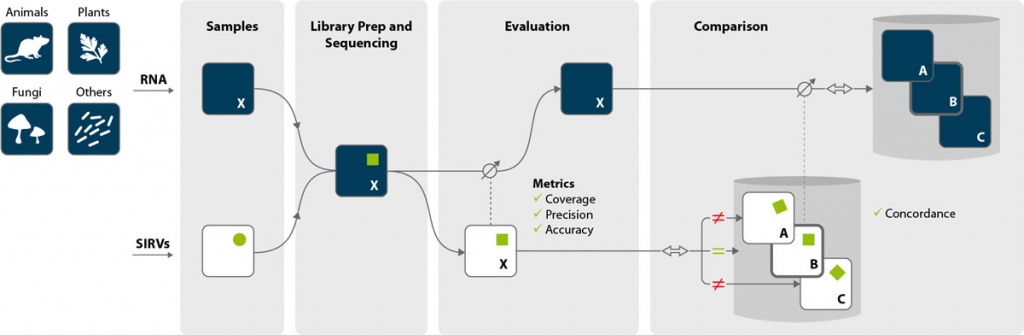
The SIRVs can be analyzed with almost any RNA-Seq protocol and any NGS platform (e.g., Illumina®, IonTorrent®, PacBio™, or Oxford Nanopore Technologies™). Being part of one sample, SIRVs undergo the very same reaction steps of library preparation and sequencing as the endogenous RNA. The sequencing data file then contains reads from SIRVs and endogenous RNA.
Evaluation
The origin of reads is determined by mapping to a combined index consisting of the reference genome and the SIRVome, the spike-in genome detailing the transcript sequences and annotations. While the SIRV data is linked to the data stemming from the endogenous RNA, it is only a fraction of its size enabling a very fast evaluation of the SIRV data subset.
The data from the SIRVs can be used for the quality control of the NGS experiment, to asses sequencing errors and biases, and for troubleshooting. The quality of RNA-Seq experiments can be determined by calculating unique quality metrics in the form of
- coefficient of deviation (CoD), calculated by comparing the measured coverage with the expected coverage,
- precision, a measure for the statistical variability, and
- accuracy, a measure of the statistical bias.
These quality metrics are derived from the spike-in transcripts but reflect the situation in the endogenous RNA data set.
Comparison
Because SIRV data sets are well defined and compact, all comparisons require proportionally less computational power ensuring fast processing. Differences between the linked SIRV data sets mirror proportionally the main data of the endogenous RNA. Concordance is independent of the accuracy but describes the coherence of data sets and identifies endogenous RNA data sets that are suitable for meaningful comparisons, e.g., for differential expression analyses.
Background
Read More
For these studies, reference RNA samples were created from Universal Human Reference RNA (UHRR from Agilent Technologies) and Human Brain Reference RNA (First Choice HBRR from Ambion, Thermo Fisher), which contain a stable but largely unknown transcript variant diversity. In addition, these reference samples contained a set of 92 in vitro transcripts as spike-in controls, which were developed by the External RNA Controls Consortium (Baker et al. 2005; Jiang et al. 2011; Munro et al. 2014). These control transcripts, ERCCs (Ambion, Thermo Fisher Scientific), allow for the assessment of dynamic range, dose response, lower limit of detection, strandedness, and fold-change response of RNA sequencing pipelines within the limitation of the monoexonic, single-isoform RNA sequences (Jiang et al. 2011). Because the ERCCs contain no transcript variants, one of the main challenges of sequencing complex transcriptomes – to identify and distinguish splice variants – could not be evaluated.
First attempts to study the quality of RNA-Seq pipelines on the transcript-isoform level were made by using mouse spike-in control transcripts, which demonstrated that abundance estimation of multiple isoform spike-ins produce lower duplicate correlations at transcript level than gene level (Leshkowitz et al. 2016). These experiments used endogenous but not expressed mouse transcripts as judged by earlier micro array measurements, making this approach time consuming, costly, and foremost not generally applicable given that each sample would require its own customized set of spike-ins. Although different bioinformatics tools were compared for adequate quantification of gene expression and transcript isoforms, a straight series of quality metrics has not been implemented for comparing results from the known controls with the unknown endogenous RNA.
The SIRV isoforms were conceived in autumn 2013, and in July 2014 the SIRV design, quality targets and production were presented and discussed at the first ERCC 2.0 workshop hosted by the National Institute of Standards and Technology Advances in Biological/Medical Measurement Science Program (NIST-ABMS) (Munro und Salit 2014). SIRVs were introduced with a test program in June 2015, and the isoform module has been commercially available since September 2015.
In September 2016, the Garvan Institute published a complementary RNA spike-in system called Sequins, also comprising naturally derived and inverted sequences. These represent on average just 2.1, and up to 4, isoforms per gene, such that 164 isoforms are distributed across 78 genes (Hardwick et al. 2016). As judged by cumulative frequency histograms the artificial gene loci correspond well to the human transcriptome structure and annotation, from which the inverted sequences were initially derived. However, the Sequins map many different features to the same RNA molecules, which hinders the systematic, unambiguous analysis of RNA-Seq pipelines and experiments. Performance at boundary conditions are difficult to resolve as Sequins are distributed across a wide concentration range of up to 6 orders of magnitude, similar to the monocistronic, single-isoform ERCCs. Errors caused by an RNA-Seq pipeline can therefore not be unambiguously attributed to difficulties caused by either too complex annotation patterns or just by low sequence coverage. Despite the large number of isoforms, the mutually exclusive exon proportion is high, and the density of multiple sequence coverage by different isoforms is low.
Within the SIRV modules, these two features – i) complex isoform features and ii) concentration gradient – are not combined but clearly separated between the SIRV isoforms and the ERCCs modules.
In July 2020, the long SIRV module was introduced, containing spike-in transcripts with lengths between 4 kb and 12 kb.


