SIRVs
The most comprehensive
RNA spike-in controls

Modular Design
Transcriptome Complexity in a Nutshell
From their conception in 2013, the Spike-In RNA Variants, SIRVs, were designed to develop into a series of modules which mimic transcriptome complexity in a condensed manner, each module probing a specific component (Figure1). Deviations of the results from the expected values can be related to specific flaws in a sample, experiment, or entire pipeline. Transcription and splicing variants were highest in demand and therefore realized first. Since the SIRV isoforms are complementary to the ERCCs, these two modules are available as a combination in SIRV-Set 3 and synergistically deliver the required information about dynamic range, lower detection limit, input-output linearity, and performance in transcript variant detection and quantification. The long SIRV module – containing 15 transcripts with lengths between 4 kb and 12 kb – was added to the portfolio in 2020 for the assessment of the length aspect of transcriptome complexity. This module is available in combination with the SIRV isoforms and the ERCCs in SIRV-Set 4.

Read more
If you are interested in participating in the design of further external RNA control modules, please contact us at info@lexogen.com, and also consider to
- post to a larger audience in the seqanswers.com SIRVs forum,
- consult relevant blogs such as CoreGenomics and RNA-Seq Blog,
- and to participate in the ERCC 2.0 consortium.
The SIRVome
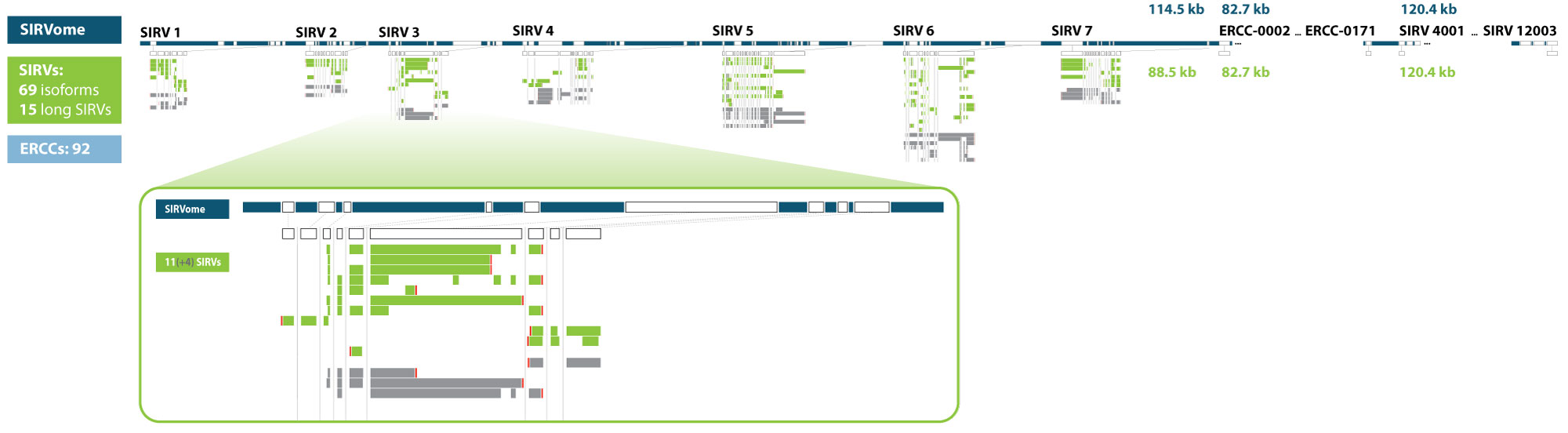
Read more
Some data analysis methods require, or benefit, from comprehensive gene definitions. These are provided for the SIRV isoforms, the ERCCs, and the long SIRVs in the annotation files, with a further 1 kB of sequences defined upstream and downstream of the first and last exon, respectively. These random sequences were created similarly to the intron sequences by mirroring the G/C content of the exon sequences and not matching to nucleotide database entries (search window 27 bp).
Isoform Module
The SIRV isoforms are a set of 69 artificial transcript variants. These were derived from 7 human model genes, and their annotated transcripts were complemented by additional isoforms to comprehensively reflect variations of alternative splicing, alternative transcription start- and end-sites, overlapping genes, and antisense transcripts. The 7 synthetic genes loci contain between 6 and 18 transcript variants each, which are on average 9.9 physically realized alternative isoforms, and more when accounting additional provisions in the annotations against which a pipeline can be tested. The SIRV isoform module is available in the form of three mixes, with molar ratios of transcripts in mixes E0, E1, and E2 at magnitudes of 0, 1, and 2, respectively.
Condensed Transcription and Isoform Complexity
The structures of seven human model genes (KLK5, LDHD, LGALS17A, DAPK3, HAUS5, USF2, and TESK2) were used as scaffold for the design of genes SIRV1 to SIRV7. The ENCODE-annotated transcripts as well as additional variants were edited to comprehensively present transcription variations like different start- and end-site usages, alternative splicing, overlapping genes, and antisense transcription.
Read more
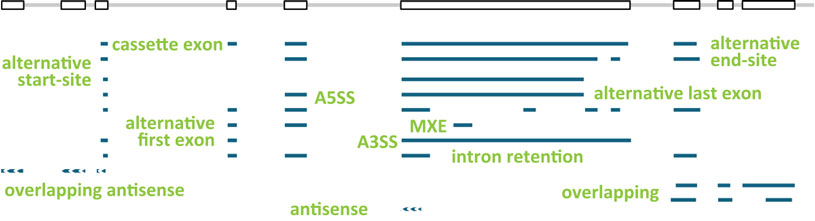
Figure 4 illustrates in one example how the human gene KLK5 served as a blue print for the design of the gene SIRV1. In addition to the 8 realized SIRV1 transcripts, 4 more were designed, which only exist in the over-annotation file provided together with the correct annotation. Vice versa, 3 transcripts of the existing SIRV1 set are not present in the insufficient annotation file. This way, transcript isoform detecting and quantifying algorithms can be challenged for their robustness towards real-life scenarios, in which the transcripts in a sample do not align with the available annotation.
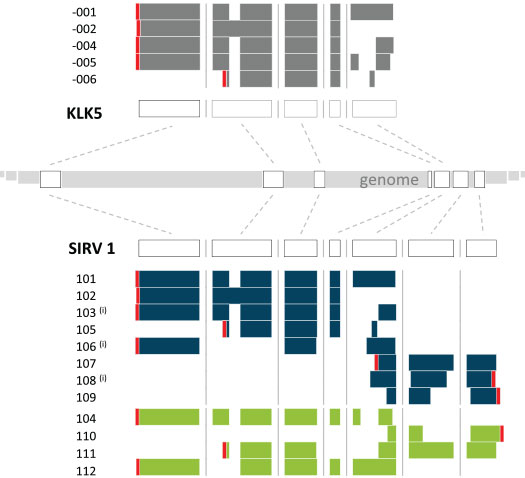
Figure 4 | Design path and exon-intron structures of the SIRV1 gene. The SIRV1 gene was derived from the human KLK5 gene, with transcripts added to the Ensembl-annotated ones to achieve a comprehensive transcriptome complexity. Transcripts in blue are part of SIRV isoform mixes, transcripts in green are only part of an over-annotation. (i) Refers to transcripts that are omitted in an incomplete annotation. The polyadenylated 3’ end is marked in red, indicating sense and antisense orientations.
Together, the 7 SIRV isoform genes model comprehensively and in a condensed and redundant manner transcription and alternative splicing variations (Table 1).
Table 1 | Summary of splice and transcription variations per SIRV isoform gene.
| Alternative 1st exon | Start site variation | Alternative 5′ splice site | Alternative 3′ splice site | Exon skipping | Exon splitting | End site variation | Alternative last exon | |
| SIRV1 | 5 | 4 | 5 | 2 | 2 | 3 | 4 | 1 |
| SIRV2 | 1 | 3 | 3 | 2 | 0 | 3 | 2 | 2 |
| SIRV3 | 1 | 5 | 5 | 4 | 5 | 4 | 7 | 4 |
| SIRV4 | 4 | 2 | 2 | 4 | 2 | 1 | 5 | 3 |
| SIRV5 | 3 | 9 | 6 | 8 | 5 | 17 | 7 | 7 |
| SIRV6 | 9 | 10 | 7 | 26 | 27 | 28 | 13 | 3 |
| SIRV7 | 2 | 5 | 1 | 1 | 31 | 1 | 4 | 3 |
The occurrences of the different events are counted for each transcript in reference to a hypothetical master transcript of maximal length containing all exon sequences from all transcript variants of a given gene. Therefore, in a formal sense no intron retention can occur, but this event is defined as exon splitting caused by the introduction of an intron sequence (illustrated in Figure 4).
The transcripts of a SIRV isoform gene are assigned to 1 of 4 SubMixes to enable preset ratios in mixes E0, E1, and E2 (see SIRV-Set 1 in SIRV Sets for more information).
Figure 5 illustrates how the known input amounts of transcripts of a SIRV isoform gene allow for precise modeling of ideal coverage expectations. RNA-Seq introduces biases to the read distribution, and hence the experimental coverage will deviate from the expected one.
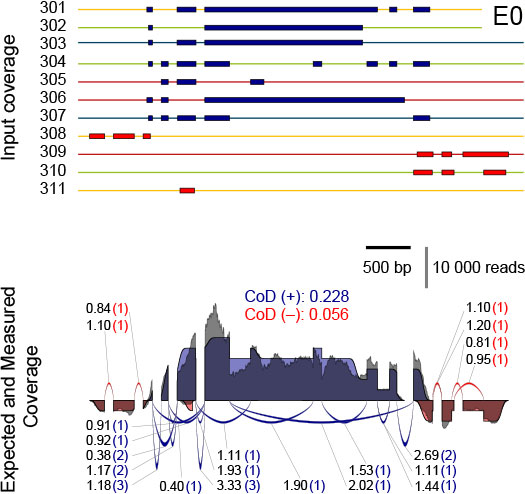
Figure 5 | Comparison of the expected and the measured coverages for the SIRV3 locus in the equimolar Mix E0. Top: individual transcripts of SIRV3 with transcripts on the plus strand in blue and in red for the ones on the minus strand. Color code indicates the SubMix allocation. Bottom: the expected SIRV3 coverage is shown as superposition of individual transcript coverages, in which the terminal sites have been modelled by a transient error function. The measured coverages after read mapping by TopHat2 are shown in grey. The measured coverages and number of splice junction reads were normalized to obtained identical areas under the curves and identical sums of all junctions for the expected and measured data. The measured splice junction reads are shown by the numbers before the brackets, while the expected values are shown inside the brackets. The CoD (Coefficient of Deviation) values are given for the plus and minus strand in the respective colors.
Native Gene Features but Unique Sequences
The SIRV isoform transcripts range in length from 191 to 2528 nt (mean 1134 nt; median 813 nt), which includes a 30 nt long poly(A)-tail. The GC-content varies between 29.5 and 51.2 % (mean 43.0 %; median 43.6 %). The exon sequences were created from a pool of database-derived genomes (gene fragments from viruses and bacteriophage capsid proteins and glycoproteins) and modified by inverting the sequence to lose identity while maintaining a naturally occurring order in the sequences.
Read more
The splice junctions conform to 96.9 % to the canonical GT-AG exon-intron junction rule with few exceptions harboring the less frequently occurring variations GC-AG (1.7 %) and AT-AC (0.6 %). Two non-canonical splice sites, CT-AG and CT-AC, account for 0.4 % each. Intron sequences that do not align with exons of another isoform were drawn from random sequences whereby the GC content was balanced to comply with the adjacent exonic sequences. The sequence exclusivity was verified by blasting the exon sequences against the entire NCBI database including ERCCs on the nucleotide and protein level. The artificial SIRV isoform sequences are suitable for noninterfering qualitative and quantitative assessments in the context of known genomic systems and complementary to the ERCC sequences (NIST SRM 2374). The SIRV isoform sequences were deposited at the NCBI’s GenBank (accession numbers KX147759 to -65 for SIRV1 to SIRV7) and can be downloaded in the Downloads section.
Certain data analysis approaches require or benefit from comprehensive gene definitions. These are provided for the SIRV isoforms in the annotation files, with a further 1 kB of sequences defined upstream and downstream of the first and last exon, respectively. These random sequences were created similarly to the intron sequences by mirroring the G/C content of the exon sequences and not matching to nucleotide database entries (search window 27 bp).
Correct, insufficient and over-annotations of SIRV isoforms
The a priori knowledge of SIRV transcript sequences and concentrations allows to assess the isoform-specific performance of an RNA-Seq experiment. In addition to the correct annotation of the SIRV isoforms, one insufficient and one over-annotation are supplied to enable the testing of NGS data evaluation algorithms for their robustness towards “real-life”, imperfect annotations (see also Figure 4). More annotations can be added to emulate situations of evolving reference annotations which accumulate transcripts discovered in samples of different origin.
ERCC Module
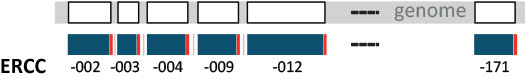
Figure 6 ǀ Single-isoform nature of ERCCs. ERCC transcripts follow the 1 gene, 1 exon, 1 transcript layout, providing each ERCC transcript with a unique sequence identity. Genes (exons) are shown in white, derived transcripts in blue, and the poly(A) tail is indicated in red to specify transcript 5’-3’ orientation. Note that while there are 92 ERCC transcripts in the mix, the RNAs are numbered non-consecutively up to 171.
The ERCCs were used in exemplary studies by the FDA Sequencing Quality Control (SEQC) Consortium and the Association of Biomolecular Resource Facilities (ABRF) (Li et al. 2014a; Li et al. 2014b; SEQC/MAQC-III Consortium 2014; Xu et al. 2014). Comparisons of the assigned and evaluated reads with known concentrations allow for the assessment of dynamic range, dose response, lower limit of detection and efficiency, as well as fold-change response of RNA sequencing pipelines, within the complexity boundaries of monoexonic, non-overlapping RNA sequences.
The RNAs are transcribed from a plasmid DNA library of ERCC sequences, available as a standard reference material from NIST (SRM 2374) (National Institute of Standards and Technology). The complete library comprises 96 unique sequences, and 92 of these were mixed in the form of transcripts assigned to four subpools with 23 ERCC controls each. Within each subpool ERCC abundances span a 220 (106) dynamic range. Similar to the SIRV isoforms, the ERCC transcripts contain a triphosphate guanosine at their 5’ end and a poly(A) tail at their 3’ end, which in the case of the ERCCs is 20-26 nt long (SIRVs; 30 nt). ERCCs on their own are available from Thermo Fisher Scientific as ERCC RNA Spike-In Mix (Cat. No. 4456740,) and ERCC ExFold RNA Spike-In Mixes (Cat. No. 4456739). The 92 ERCC sequences are available in the Downloads section.
Long SIRVs Module
The introduction of long read sequencing platforms like Pacific Biosciences™ and Oxford Nanopore Technologies™ has significantly increased the available read length, now easily exceeding the average transcript length. The ERCC and SIRV isoform modules are optimized for assessing RNA abundance and isoform complexity aspects. However, the average ERCC length is 909 nt (max. 2036 nt), the average SIRV isoform length is 1134 nt (max. 2528 nt), and thus spike-in RNA transcripts of both modules are below the average reported length for eukaryotic protein-coding mRNAs (e.g. 3.5 kb for the human transcriptome (Piovesan et al., 2019)).
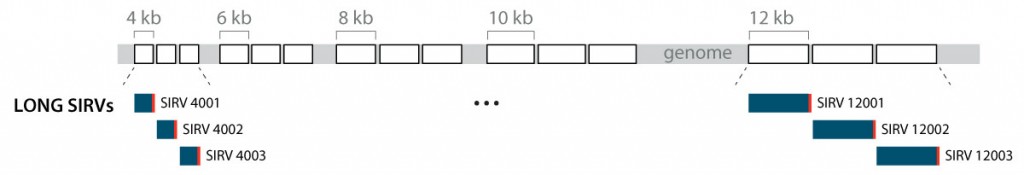
Lexogen has therefore developed “long SIRVs”, a module that contains three different transcripts for each of the five length categories 4 kb, 6 kb, 8 kb, 10 kb, and 12 kb (Figure 7). These RNAs cover the length of the majority of cellular transcripts. The sequence of each of these 15 RNAs is unique and does not overlap with any other spike-in or endogenous transcripts (similar to the ERCC module). Therefore, the equimolar long SIRVs are optimal tools to evaluate the transcript length aspect in RNA-Seq workflows. While designed in particular to assess long-read platforms, long SIRVs reveal length dependencies also in short read workflows.
Sequence and annotation
Certain data analysis approaches require or benefit from comprehensive gene definitions. These are provided for the long SIRVs in the annotation files, with a further 1 kB of sequences defined upstream and downstream of the first and last exon, respectively. These random sequences were created mirroring the G/C content of the exon sequences and not matching to nucleotide database entries (search window 27 bp). The long SIRV sequences can be downloaded in the Downloads section.


