Small RNA-Seq Library Prep Kit
- Gel-free user-friendly workflow
- Multiplexing of up to 96 samples
- Ready-to-sequence libraries in less than 5 hours
- Wide input range from 50 pg to 1,000 ng RNA
- Optimized for low RNA content samples such as plasma, serum, and urine
Description
Small RNA-Seq Library Prep Kit for Illumina
The Small RNA-Seq Library Prep Kit provides a protocol for generating small RNA libraries for Illumina sequencing directly from total RNA or enriched small RNA.
Multiplexing of up to 96 Samples
Multiplexing of up to 96 samples is possible with complimentary i7 indexes provided in the kit. This allows you to pool more samples per sequencing lane and perform cost-efficient experiments on the platforms of different scale – from bench top to high throughput instruments.
Gel-free User-Friendly and Fast Protocol
Lexogen’s Small RNA-Seq kit offers a time saving protocol that can be completed within 5 hours, and requires just about 1 hour of your hands-on time. The final library does not need to go through gel purification. Quick and convenient magnetic bead-based purification can optionally be performed to remove adapters dimers for some demanding RNA inputs.
High Reproducibility
The protocol exhibits exceptional reproducibility and correlation across samples with the different concentrations (Figure 1).

Exceptional miRNA Discovery
The protocol allows for detection of a higher number of microRNAs than other workflows. The difference is especially considerable for lower RNA inputs (Figure 2).

Optimized for RNA from Challenging Sources
The high sensitivity of the protocol makes it very well suited for challenging, low content RNA sources, such as liquid biopsies (plasma, serum, and urine), including exosomes. The protocol has also been tested on RNA from cells and tissues. The kit can be used for inputs from 100 ng – 1,000 ng of cellular total RNA or 50 pg – 1,000 ng enriched small RNA including plasma, serum, and urine.
Lexogen’s Complete Solution for Small RNA Analysis
Lexogen’s SPLIT RNA Extraction Kit offers the opportunity to extract NGS-grade quality total RNA or small and large RNA fractions from the same sample. SPLIT is highly suitable for purification of total RNA and enrichment for small RNA for use with the Small RNA-Seq Library Prep Kit. Thus, the combination of these two protocols offers you the complete solution for small RNA sample preparation for sequencing on any of the Illumina platforms.
Workflow
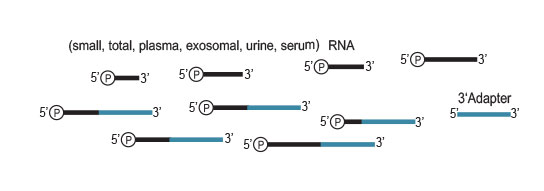
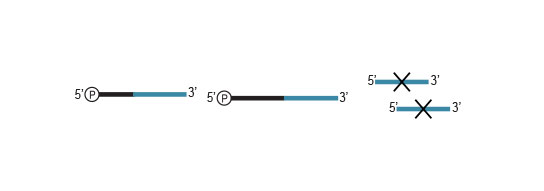
Excess amount of 3’ Adapter is removed by column purification.
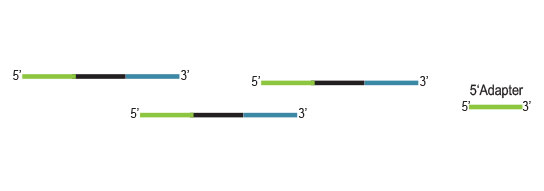
5’ Adapter ligation is taking place.
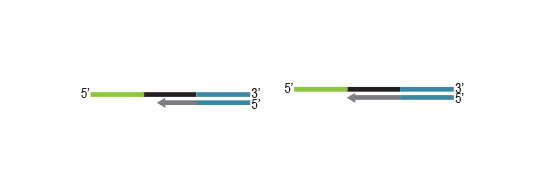
The input RNA, flanked by 5’ and 3’ Adapters is converted into cDNA.
And double stranded libraries are produced.
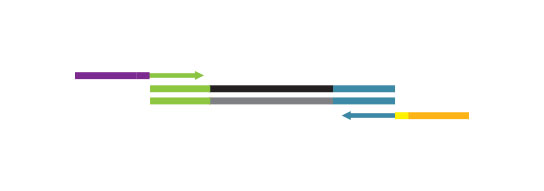
Multiplexing indices are introduced during the PCR amplification step.

The library product is then subjected to a clean-up and concentration step.

Libraries are ready to be sequenced on an Illumina platform.
FAQ
Frequently Asked Questions
Access our frequently asked question (FAQ) resources via the buttons below.
Please also check our General Guidelines and FAQ resources!
How do you like the new online FAQ resource? Please share your feedback with us!
Downloads
Safety Data Sheet
If you need more information about our products, please contact us through support@lexogen.com or directly under +43 1 345 1212-41.
Data Analysis
Bioinformatics Services: Leave the analysis to the RNA experts
We offer a wide range of bioinformatics services ranging from standard analysis to fully custom data analysis projects. Our bioinformatics team consists of data analysis experts with experience in various NGS data analysis pipelines who are passionate about developing novel, customized workflows and solutions while keeping biology at the forefront.
Analyzing data on your own?
Check our tips and guidelines in FAQs for Small RNA.
For more information, please contact us at support@lexogen.com.
First time user of Small RNA-Seq?
First Time User? We’re excited to offer you an exclusive introductory offer.
Buy from our webstore
Need a web quote?
You can generate a web quote by Register or Login to your account. In the account settings please fill in your billing and shipping address. Add products to your cart, view cart and click the “Generate Quote” button. A quote in PDF format will be generated and ready to download. You can use this PDF document to place an order by sending it directly to sales@lexogen.com.
Web quoting is not available for countries served by our distributors. Please contact your local distributor for a quote.



