The Spike-In RNA Variants of the isoform, the ERCC, and the long SIRV modules (see Modular Design) are realized as defined mixes, and for some modules, different mixes are available. Mixes, and combinations thereof, are available in the form of SIRV sets.
We use the following definitions:
Module
Group of spike-in controls that mimic predominantly one aspect of transcriptome complexity.
Mix
SIRVs of the same module that are combined in precise defined molarity.
Set
Term for the combination of mixes or modules.
Table 1 presents a SIRV set selection guide to help you finding the right set of modules and mixes for your application.
Table 1 ǀ SIRV set selection guide.
SIRV-Set 1
SIRV-Set 2
SIRV-Set 3
SIRV-Set 4
Cat. No
025.03
050.0*
051.0*
141.0*
Module(s)
Isoforms
Isoform Mixes E0, E1, E2
Isoform Mix E0
Isoform Mix E0
Isoform Mix E0
ERCC
X
X
ERCC Mix 1
ERCC Mix 1
long SIRVs
X
X
X
long SIRVs
Property
Isoform detection, and quantification
✓
✓
✓
✓
Dynamic range
partially
X
✓
✓
Length > 2.5 kb
X
X
X
✓
Applications
Pipeline Validation
✓
partially
partially
partially
Sample Control
X
✓
✓
✓
Number of spike-intranscripts in each mix
69 (69 isoforms in each Mix)
69 (69 isoforms)
161 (69 isoforms, 92 ERCCs)
176 (69 isoforms, 92 ERCCs, 15 long SIRV)
SIRV-Set 1 (Cat. No 025) contains the isoform mixes E0, E1 and E2 of the isoform module, SIRV-Set 2 (Cat. No 050) provides the isoform Mix E0 only, SIRV-Set 3 (Cat. No 051) has the SIRV Isoform Mix E0 in a mixture with the ERCCs, and SIRV-Set 4 (Cat. No 141) is a mixture of the long SIRVs with SIRV Isoform Mix E0 and the ERCCs.
*Refers to number of vials, 1 or 3. The ERCC Module includes ERCC Mix 1 (Munro et al., 2014) ✓: applicable, X : not applicable, and partly applicable (or parts of the sets applicable).
For more information please consult the respective User Guides in the Downloads section.
SIRV-Set 1: Isoform Mixes E0, E1 and E2
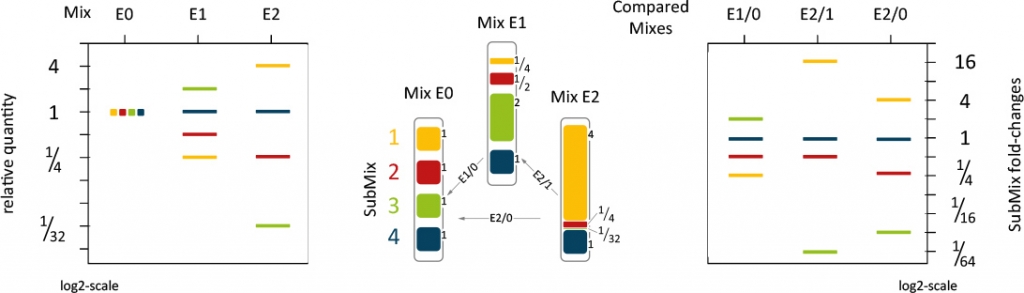
The isoform module is available in SIRV-Set 1 (Cat. No. 025) in 3 SIRV mixes, termed E0, E1 and E2, with each mix containing all 69 SIRV isoform transcripts (from 7 SIRV genes) but in different concentration ratios. E0 is ideal for assessing the detection capabilities of a given RNA-Seq workflow, since all 69 transcripts are present in equimolar concentrations, and their detection should be unbiased and not a function of read depth or similar. E1 already contains a moderate concentration distribution of transcript variants of a given gene, and E2 represents the natural situation, whereby a dominant, abundant transcript variant is transcribed from a gene together with (up to 17) other variants present at lower expression levels (down to <1%). The latter situation is already quite challenging for correct transcript determination based on short read assembly but also tests efficiently the linearity and sensitivity of long-read sequencing platforms and protocols that cannot rely on millions of reads (Figure 1).
Figure 1 ǀ Distribution of the 4 SubMixes in the 3 isoform mixes and the resulting intra- and inter-mix ratios. Left, the intra-mix concentration ratios provide three different concentration settings to evaluate accuracy in relative concentration measurements. Right, the present fold-changes allow for 3 possible inter-mix comparisons to evaluate differential gene expression measurements. SubMixes 1-4 are indicated by their respective colors, and transcript isoforms of each of the 7 SIRV genes are distributed across all SubMixes.
Remarkably, by comparing the SIRV transcript quantifications in different mixes (E0 vs E1, E0 vs E2, or E1 vs E2) differential gene expression can be evaluated on the transcript isoform and variant level. It can be assessed, if biases in transcript detection are similar for both mixes tested and therefore having a significant effect on transcript quantification in a given mix, while having no or only a limited effect on differential transcript/gene expression quantification. Combining the individual SIRV transcript expression quantifications yields a value for SIRV gene expression, and the accuracy of this evaluation might differ significantly from the deviations seen on the transcript level.
SIRV-Set 2: Isoform Mix E0
The isoform Mix E0 is available on its own as SIRV-Set 2 (Cat. No 050) with all 69 isoforms being present at equimolar concentrations. Its applications include RNA-Seq experiments that need to be validated for the detection of a complex mixture of isoforms without applying a high read depth to cover transcripts at different concentrations. Among these are sequencing runs on long-read NGS platforms as provided by Oxford Nanopore Technologies and Pacific Biosciences. These can produce full-length reads to identify the isoforms faithfully. However, unlike short-read platforms they do not provide the millions of reads necessary to detect and quantify isoforms across a larger dynamic range, in particular if these spike-ins only constitute a very small fraction of the total RNA.
Deviations from the expected equimolar outcome can be quantified, which allows for evaluation of the performance of isoform-centered workflows. On the gene level, quantifications of the individual SIRV isoform can be summed up for each SIRV gene, which permits the validation of pipelines working with data stemming from individual isoforms but focused on gene expression calculations only.
SIRV-Set 2 is very suitable for the calculation of concordance, since the experiments’ fingerprints depends solely on their dealing with the SIRV isoform’s complexity but not on input concentration differences between these isoform transcripts.
Reads from SIRV-Set 2 can be downsampled to emulate data representing the situations in lower concentration ranges. A repeated mapping and assignment provides adequate measures for the RNA-Seq experiment ability to detect variants and measure its concentrations in a different band with. Using such iterative approach SIRV-Set 2 is capable to map the entire abundance spectrum.
SIRV-Set 3: Isoform Mix E0 & ERCCs
SIRV-Set 3 (Cat. No. 051) contains the isoform Mix E0 and the ERCC Mix 1 in equal shares. Both contribute equally to the final mass.
The mixture of 69 SIRV isoform transcripts and 92 non-overlapping ERCC RNAs addresses the need for complex spike-in RNA controls that cover both, a high level of isoform complexity and a large concentration range. Together, they enable an even more comprehensive quality assessment and monitoring across the whole RNA-Seq workflow to derive technical details and telling fingerprints for comparing individual samples, and experiments.
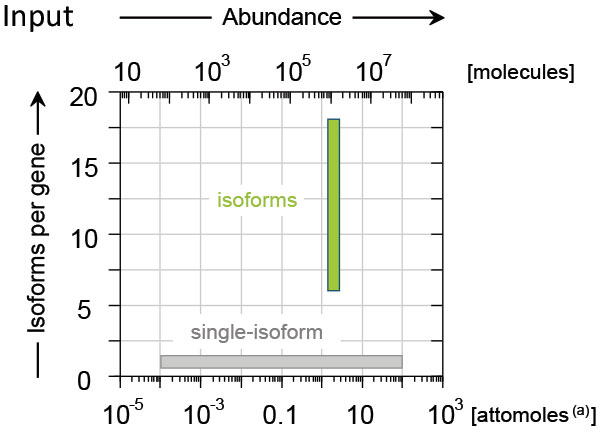
The single-isoform ERCC transcripts cover concentrations of 6 orders of magnitude and are complemented by the equimolar SIRV isoforms. Figure 2 illustrates this added dimension by showing the covered complexity plotted versus the input concentration.
Figure 2 ǀ Concentrations and complexity of SIRV isoforms and single-isoform ERCC transcripts in SIRV-Set 3. Top; the isoform module with 69 transcripts in 7 gene loci contains all species at the same molarity (green bar). It covers the medium to high range of natural occurring isoform complexity. The single-isoform module with 92 ERCC transcripts covers concentrations of 6 orders of magnitude (grey bar), which is sufficient to represent the entire dynamic range of natural occurring transcripts. (a) The amount of attomoles refers to the typical amount that is spiked into 100 ng total RNA with the aim of attracting approx. 1% of the mRNA-Seq reads – subject to mRNA content and pipeline parameter for which, of course, the modules control for.
The accuracy (systematic error) and precision (random error) in quantifying single-isoform transcripts in RNA-Seq experiments is predominantly concentration- and read-depth dependent. While reads usually map uniquely to ERCC transcripts, the precision remains coverage dependent with reads following typically a Poisson distribution.
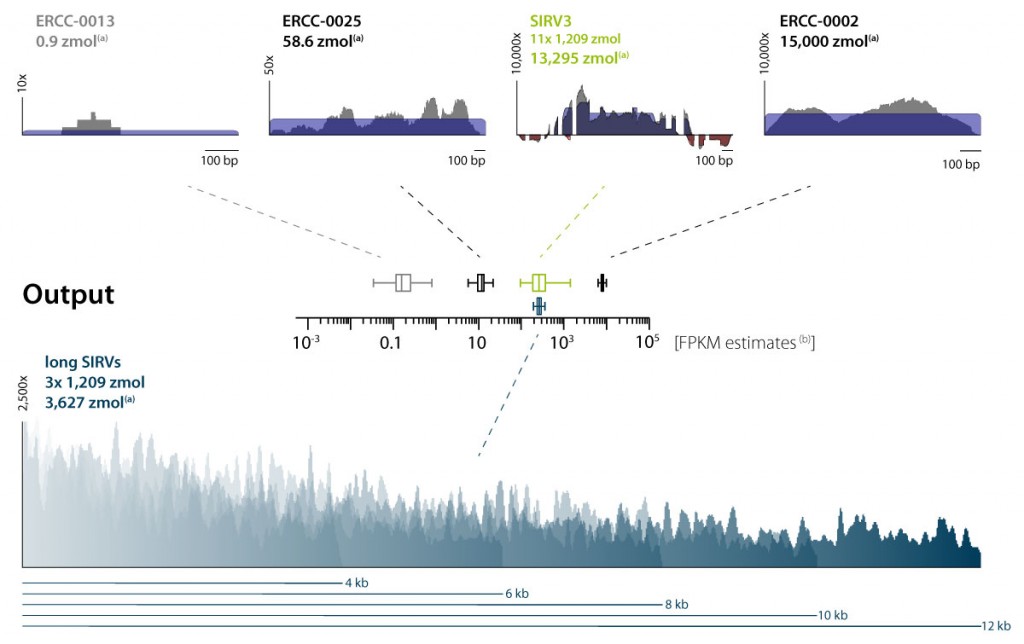
Isoform detection and quantification requires a sufficient coverage, and therefore the isoform spike-ins are added all at the same concentration and in the upper range of the ERCCs. Thereby, the issue of identifying a given isoform is not mingled up with differing concentrations. The overall higher input concentration allows for sufficient reads to be obtained in RNA-Seq experiments for isoform identification. Still, quantification of SIRV isoforms remains challenging on both short-read systems (due to assignment issues) and long-read platforms (e.g. because of per base error, low read numbers, and amplification bias). This is indicated in Figure 3 by a larger error margin for the isoforms than e.g. for ERCCs at an even lower concentration.
Figure 3 ǀ Read coverages of SIRV isoform and single-isoform (ERCC) genes depend on input concentration, library preparation efficiency, biases, and read depth. Quantifying the 92 single-isoform transcripts (ERCCs) depends on the averaged overall coverage but is rather independent of positional coverage fluctuations. Three ERCC examples of different input abundance are shown. With 6-18 isoforms mapping to the 7 SIRV gene loci, NGS read assignment and subsequent isoform quantification is much more challenging and depends strongly on coverage uniformity. One gene with 11 SIRV isoforms is shown alongside the ERCCs. The blue areas represent the expected coverage in the sense direction, and the red areas the expected coverage in the antisense direction. The grey areas show exemplary coverages from one stranded library preparation that has been sequenced in paired end mode. (a) The number of zettamoles refers to the total amount per SIRV-Set 3 vial. (b) Reflects the FPKM bandwidth of the controls when those occupy around 1 % of the reads in an mRNA-Seq experiment. The long SIRVs are present at concentrations identical to the SIRV isoforms, but quantification is not affected by isoform complexity, resulting in a smaller standard error. The lower panel shows the read coverage of all long SIRVs as a result of sequencing CORALL™ total RNA-Seq libraries. Coverages are averaged for each length category.
SIRV-Set 4: Isoform Mix E0, ERCCs, and long SIRVs
SIRV-Set 4 (Cat. No. 141) contains the long SIRV module with 15 RNAs of 4 – 12 kb length in addition to the 69 isoforms and 92 ERCC transcripts of SIRV-Set 3. The long SIRVs are present at concentrations identical to the SIRV isoforms, and therefore quantification is not affected by isoform complexity, resulting in a smaller standard error (Figure 3).
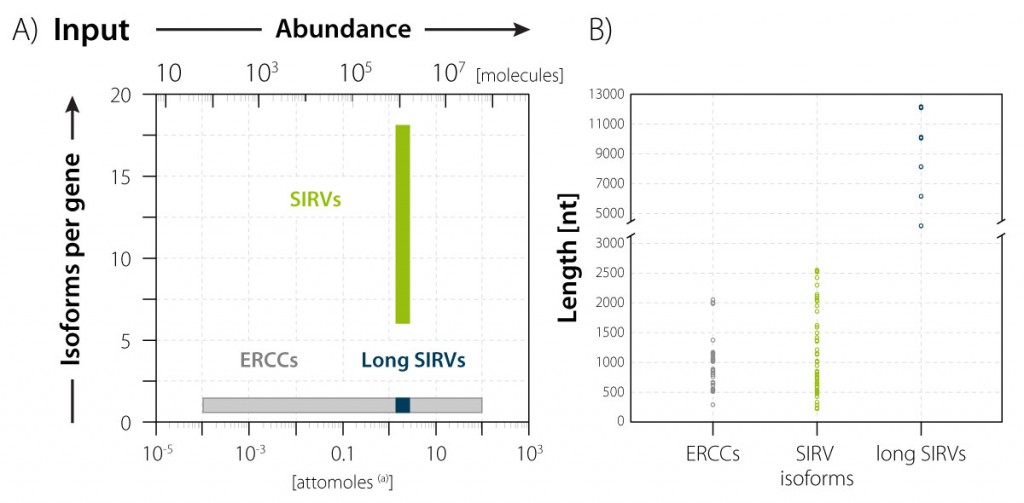
SIRV-Set 4 covers three spike-in aspects: isoform complexity, abundance, and length. These are clearly segregated with the long SIRVs having non-overlapping sequences and equal concentrations (Figure 4).
Figure 4 | A) Isoform and abundance complexity and B) Length complexity in SIRV-Set 4. The SIRV isoform and ERCC transcripts in SIRV-Set 3 control for the two main dimensions of transcriptome complexity: isoforms and abundance. SIRV-Set 4 additionally contains controls for length complexity. The isoform module with 69 transcripts from 7 genes contains all species at the same molarity (green bar). The single-isoform module with 92 ERCC transcripts spans a concentration range of 6 orders of magnitude (grey bar), which is sufficient to cover the entire dynamic range of naturally occurring transcripts. (a) The amount of attomoles refers to the typical amount that is spiked into 100 ng total RNA with the aim to obtain approx. 1 % of the mRNA-Seq reads. Long SIRVs (blue bar) contain 1 transcript per gene and are present at equimolar concentrations in SIRV-Set 4. B) Transcripts of the ERCC module range up to 2 kb in length, the ones of the SIRV isoform module up to 2.5 kb. The long SIRV module contains three transcripts in each of the length categories 4 kb, 6 kb, 8 kb, 10 kb and 12kb.
Cookie Settings
By clicking on "Accept All" you allow us to provide personalized content and ads, analyse usage statistics, and improve site functionality. Click "Accept All" to consent to these uses or click "Configure" to manage your cookie settings. You may change your cookie settings at any time.
Please choose which cookies you'd like to use.
Required
These cookies are required to provide core site functionality. These can't be disabled.
Analytics
These cookies allow us to analyse usage of our site so we can improve its performance.
Marketing
These cookies are used by advertisers to show you ads relevant to your interests.