Access Alternative Polyadenylation with ease
– QuantSeq REV V2
Do you have any questions?
Find the ideal kit for your application:
QuantSeq 3’ mRNA-Seq V2 Library Prep Kit REV
The QuantSeq REV Kit generates Illumina-compatible 3’ mRNA-Seq libraries directly targeting the 3’ end of polyadenylated transcripts. The first nucleotide of the read- out corresponds to the genuine transcription end site. The combination of cost-efficient 3’ mRNA sequencing and authentic end site representation makes QuantSeq REV the perfect tool to investigate alternative polyadenylation sites for projects at any scale.
Alternative Polyadenylation (APA)
(noun) / ȯl-ˈtər-nə-tiv päl-ē- -ə-ˈden-ə- lā shən
Alternative polyadenylation (APA) is a fundamental mechanism in the regulation of eukaryotic gene expression. Unlike traditional polyadenylation at the canonical 3’ end of a transcript, APA generates distinct mRNA isoforms through selection of alternate polyadenylation sites located at a different position within the 3’ end of the transcript or even in another exon. Alternative 3’ ends impact transcript properties and expression levels and are of utmost importance for diverse biological processes, e.g., development and disease. By investigating the dynamics of APA, we deepen our comprehension of cellular regulation, which opens new avenues for exploring potential therapeutic interventions.
Applications
- Alternative polyadenylation analysis
- Precise transcript end site mapping
- Gene expression analysis
Performance
Robust Performance over a Wide Range of Inputs
QuantSeq REV has a broad input range of 1 ng – 500 ng total RNA without the need for any prior poly(A) selection or ribodepletion and reliably delivers high-quality data and excellent gene detection rates (Tab. 1).
Table 1 | Mapping stats for QuantSeq REV libraries across different input ranges of Universal Human Reference RNA (UHRR). Libraries were sequenced in paired end mode at 500 K reads and Read 2 was analyzed, averages over at least two replicates per experiment are presented.
| Input RNA amount | |||
|---|---|---|---|
| 500 ng | 10 ng | 1 ng | |
| % Reads Mapped | 98.9 % | 98.6 % | 98.0 % |
| % Uniquely Mapping Reads | 91.9 % | 90.0 % | 88.8 % |
| % Protein Coding | 95.5 % | 94.6 % | 93.4 % |
| No. of Genes Detected | 14,215 | 14,274 | 9,922 |
Analysis of Low Input and Low Quality Samples
The required input amount of total RNA is as low as 1 ng. QuantSeq REV is suitable to reproducibly generate libraries from low quality RNA, including FFPE samples.
Mapping of Transcript End Sites and Alternative Polyadenylation Sites
QuantSeq REV allows to exactly pinpoint the 3’ end of poly(A) RNA and to obtain accurate information about a transcripts’ 3’ UTR, including alternative polyadenylation events (Fig. 1. Tab. 2).
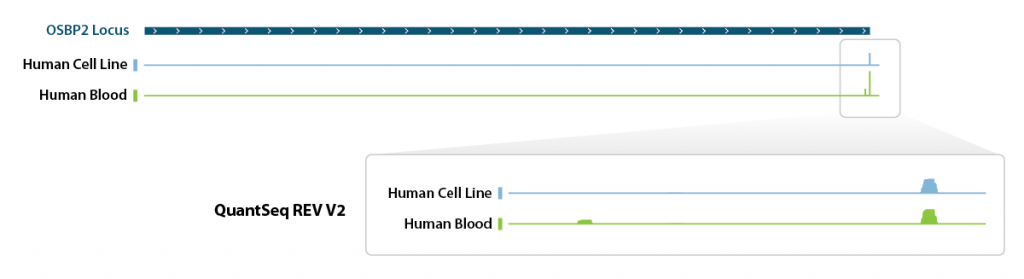
Table 2 | Alternative poly(A) site detection in samples derived from human cell lines vs. whole blood.
| All | Significant* | |
|---|---|---|
| No. of Genes with alternative poly(A) sites | 1896 | 118 |
| Known alternative poly(A) sites detected | 1894 | 118 |
| Novel alternative poly(A) sites detected | 94 | 1 |
*Significance filtering using DEXSeq v1.44.0 at padj <0.1
Libraries were generated with QuantSeq REV V2 and sequenced with a read depth of 2 – 2.5 M reads / sample. Alternative poly(A) site analysis was conducted by peak calling after filtering internally poly(A)-priming reads, removal of low abundance peaks (<5 reads) and removal of intergenic, antisense peaks, and peaks associated with overlapping genes.
Need help with your data analysis? Lexogen offers alternative poly(A) site analysis as part of our Lexogen Bioinformatics Services portfolio. Find out more!
Deplete Globin mRNA during QuantSeq Library Prep
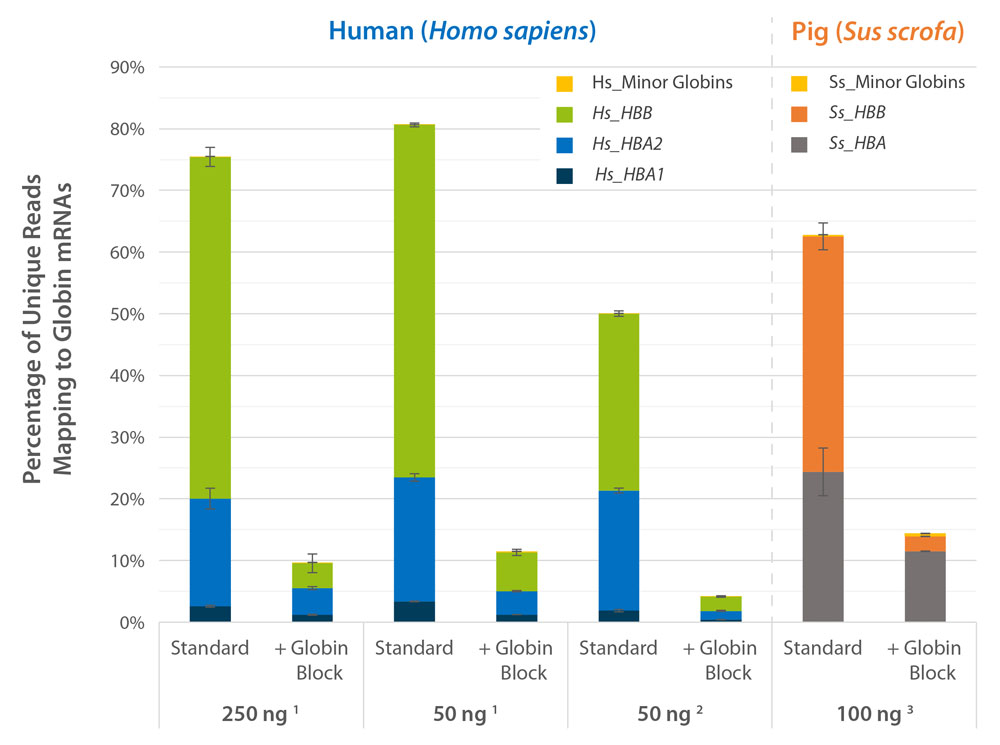
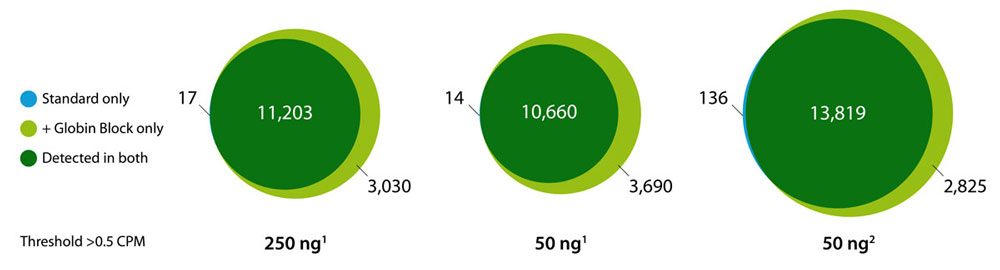
Globin Block Modules for QuantSeq enable the generation of globin-depleted, ready-to-sequence 3’ mRNA-Seq libraries from as little as 50 ng of total RNA from whole blood without additional processing steps. Globin Block reduces QuantSeq reads mapping to globin mRNAs from 80 % down to 5 % (Fig. 2) thereby dramatically increasing gene detection (Fig. 3).
Deplete Abundant Transcripts in Rodent Brain Samples with BC1 Block and QuantSeq
The BC1 Block Module (RS-BC1 Block) for QuantSeq prevents the generation of library fragments from the abundant BC1 transcripts that are present in rodent brain samples by blocking their extension during second strand synthesis.
High Strand-Specificity
QuantSeq maintains exceptional strand-specificity of >99.9 % and allows to map reads to their corresponding strand on the genome, enabling the discovery and quantification of antisense transcripts and overlapping genes.
Rapid Turnaround
QuantSeq’s simple workflow allows generating ready-to sequence NGS libraries within only 4.5 hours, including less than 2 hours hands-on time.
Direct Counting for Gene Expression Quantification
Just one fragment per transcript is produced; therefore no length normalization is required. This allows more accurate determination of gene expression values and renders QuantSeq the best alternative to microarrays and conventional RNA-Seq in gene expression and eQTL (expression quantitative trait loci) studies.
Cost Saving Multiplexing
QuantSeq REV libraries are intended for a high degree of multiplexing. The kit includes 24 or 96 Unique Dual Indices (UDIs) with an unprecedented error correction accuracy. Up to 384 UDIs are compatible with QuantSeq REV. For multiplexing needs beyond 384 samples, please consult with us at support@lexogen.com.
Lexogen’s Automated Data Analysis Solution
Lexogen offers an automated solution for QuantSeq data analysis based on state-of-the-art Lexogen’s proprietary pipeline. Thus, every user, even without bioinformatics experience, can analyze QuantSeq samples in a convenient and fast way. More information and details about Lexogen’s data analysis pipeline can be found here.
Workflow
QuantSeq has a short and simple workflow and can be completed within 4.5 hours. The required hands-on time is less than 2 hours. The kit uses total RNA as input, hence no prior poly(A) enrichment or rRNA depletion is needed.

The kit uses total RNA as input, hence no prior poly(A) enrichment or rRNA depletion is needed.
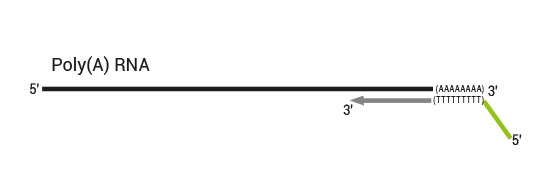
Library generation starts with oligodT priming containing the Illumina-specific Read 1 linker sequence.
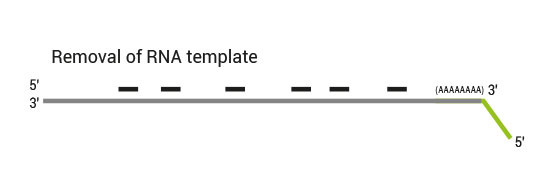
After first strand synthesis the RNA is removed.
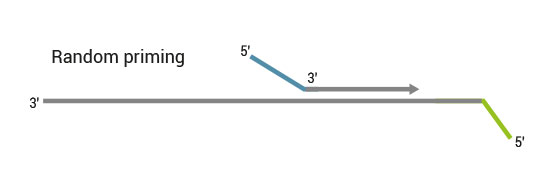
Second strand synthesis is initiated by random priming and a DNA polymerase. The random primer contains the Illumina specific Read 2 linker sequence.
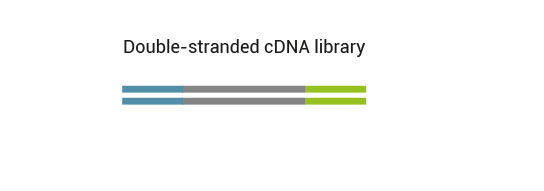
No purification is required between first and second strand synthesis. Second strand synthesis is followed by a magnetic bead-based purification step rendering the protocol compatible with automation.
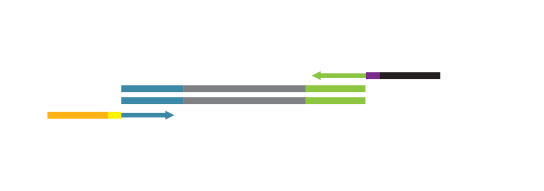
During the library amplification step sequences required for cluster generation are introduced.
Multiplexing can be performed with up to 9,216 barcode combinations using the 96 available i7 indices and 96 i5 indices.
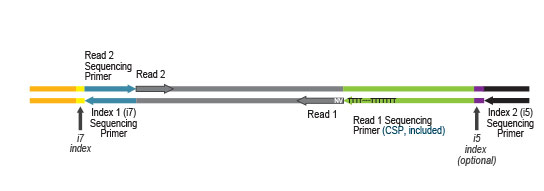
NGS reads are started at the very last nucleotide of the mRNA using the Custom Sequencing Primer (CSP Version 2, included in the kit) for sequencing. The reads generated during Read 1 reflect the cDNA sequence. With QuantSeq REV also paired-end sequencing is possible.
Data Analysis on Kangooroo
Each QuantSeq REV kit includes a voucher code for complimentary data analysis on Kangooroo - Lexogen’s web-based interactive RNA-Seq analysis platform. Analyse your data quickly and conveniently - no coding skills are required.
Featured Publications
Automation
Automated QuantSeq protocol for 3’ mRNA-Seq Library Preparation
Automating the process of library preparation has the advantage of avoiding sample tracking errors, dramatically increasing throughput, and saving hands-on time.
QuantSeq library preparation and has been successfully implemented on several liquid handlers:
- Perkin Elmer: Sciclone® / Zephyr®
- Hamilton: Microlab STAR / STARlet
- Agilent: NGS Workstation (NGS Bravo Option B)
- Beckman Coulter: Biomek FXP, Biomek i5, Biomek i7
- Eppendorf: EpMotion® 5075
- Opentrons® OT-2
QuantSeq automation on other platforms may also be possible. Please contact support team for more information.
Two key parameters must always be considered when automating a protocol:
- volume optimization
- script compatibility
In some instances, our kits will provide enough reagents while, in other instances, you will need a higher reagent volume or script adjustments. At Lexogen, we will help you find the best solution, tailored to your needs.
Before starting any new project involving automation, please reach out to our experts at support@lexogen.com
Please, also consider liaising with the robotic platform support team – they will be able to share the most recent version of the script.
Lexogen gladly supports the implementation of Lexogen-manufactured kits on liquid handlers, but not hardware or software issues linked to the original liquid handling instrument supplier.
Related resources:
FAQ
Frequently Asked Questions
Access our frequently asked question (FAQ) resources via the buttons below.
Please also check our General Guidelines and FAQ resources!
How do you like the new online FAQ resource? Please share your feedback with us!
Downloads
QuantSeq 3′ mRNA-Seq Library Prep Kit REV for Illumina
Safety Data Sheet
If you need more information about our products, please contact us through support@lexogen.com or directly under +43 1 345 1212-41.
QuantSeq Bioinformatics Data Analysis
Find more about the QuantSeq Data Analysis here.
Ordering information
| Cat. No. | Product Name |
| 225.24 | QuantSeq 3’ mRNA-Seq V2 Library Prep Kit (REV) with Custom Sequencing Primer and UDI 12 nt Set B1, 24 preps |
| 225.96 | QuantSeq 3’ mRNA-Seq V2 Library Prep Kit (REV) with Custom Sequencing Primer and UDI 12 nt Set B1, 96 preps |
First time user of QuantSeq 3’ mRNA-Seq?
First Time User? We’re excited to offer you an exclusive introductory offer.
Buy from our Webstore
Need a web quote?
You can generate a web quote by Register or Login to your account. In the account settings please fill in your billing and shipping address. Add products to your cart, view cart and click the “Generate Quote” button. A quote in PDF format will be generated and ready to download. You can use this PDF document to place an order by sending it directly to sales@lexogen.com.
Web quoting is not available for countries served by our distributors. Please contact your local distributor for a quote.



