The right choice for large scale, high-throughput RNA-Seq projects
– QuantSeq-Pool
Do you have any questions?
Find the ideal kit for your application:
QuantSeq-Pool Sample-Barcoded 3′ mRNA-Seq Library Prep Kit
QuantSeq-Pool is the optimal solution for gene expression profiling for large screening projects using sample barcoding, early pooling, and batch processing of up to 96 samples in one reaction providing a workflow that is easily scalable for multiplexing up to 36,864 samples.
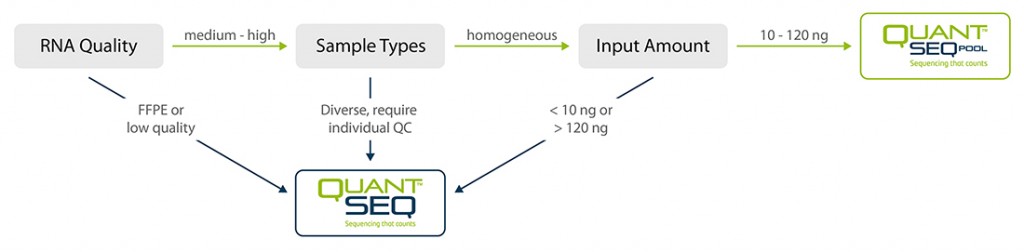
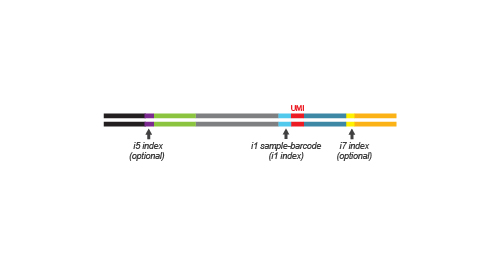
QuantSeq-Pool is the newest addition to the QuantSeq product line combining the benefits of the well-established QuantSeq methodology with sample barcoding and early pooling. Due to the convenient workflow and scalable multiplexing capacities, QuantSeq-Pool is the optimal choice for completing gene expression profiling studies in the most cost- and time-efficient way. Not sure whether QuantSeq-Pool is the right version of QuantSeq for you? Please check the QuantSeq selection guide to find out (Fig. 1).
Performance
Increased Consistency and Reduced Technical Variability
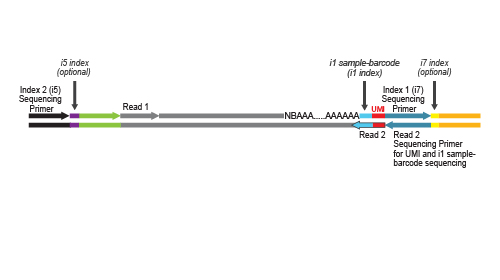
QuantSeq-Pool contains the Illumina Read 1 linker sequence in the second strand synthesis primer, hence NGS reads are generated towards the poly(A) tail, directly reflecting the mRNA sequence. Sample-barcodes and Unique Molecular Identifiers (UMIs) are introduced in the first step and are accessible in Read 2 (see workflow). Early pooling and batch processing reduce technical variation in the complete workflow and allow consistent processing of up to 96 samples in parallel. Excellent reproducibility between replicates is shown in Fig. 2.

Figure 2 | Excellent correlation between replicates for individual samples in QuantSeq-Pool. Libraries were prepared from 10 ng UHRR, sequenced, and correlated on gene level. R² was calculated by orthogonal regression.
Robust Gene Detection at Low Sequencing Depth
QuantSeq-Pool reliably detects 7,500 to 9,000 highly expressed genes at very shallow read depths of 100 K to 1 M reads per sample (Fig. 3).
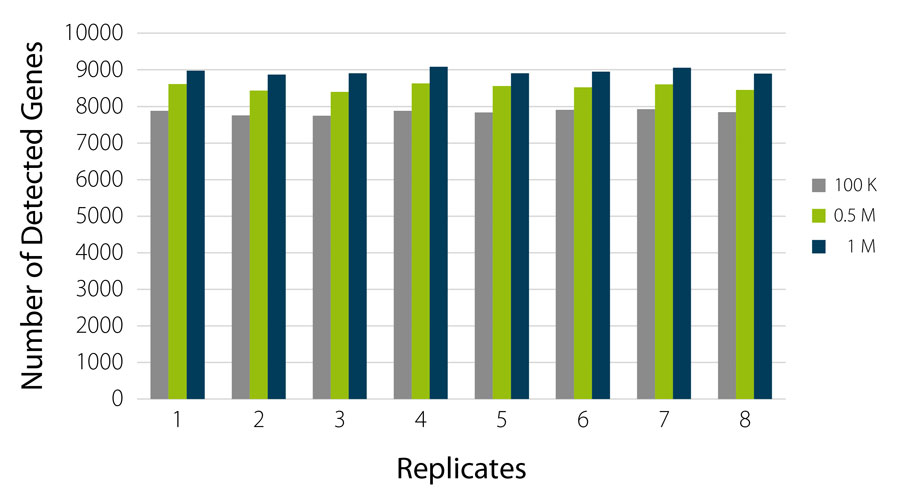
Figure 3 | QuantSeq-Pool enables robust and consistent gene detection already at low sequencing depth. Libraries were generated from 8 x 10 ng UHRR, sequenced, and gene detection was analyzed for 100 K, 0.5 M and 1 M reads / sample. Number of detected genes was counted at a threshold of > 10 Counts Per Million (CPM).
Cost Saving Multiplexing
QuantSeq-Pool libraries are intended for a high degree of multiplexing: 96 sample-barcodes (i1 indices) are included in the Kit (Cat. No. 139). Additional i5 and i7 indices are required for multiplexing more 96 samples for sequencing. Lexogen UDI 12 nt Sets with pre-mixed i5 and i7 indices offer superior error correction capacity and are recommended for massive multiplexing (Cat. No. 101 – 105, 156). Lexogen’s indices are provided in a convenient 96-well format for multiplexing of up to 9,216 samples per lane for each 96-well index plate. Lexogen UDI 12 nt Unique Dual Indexing Sets are available for up to 384 12 nt UDIs increasing the multiplexing capacity to 36,864 samples per sequencing lane. For further indexing options, please contact support@lexogen.com.
This high level of multiplexing allows decreasing costs by:
- Saving sequencing space
- Rescuing most of the reads (error correction – best when used with 12 nt UDI)
- Avoiding misassigned reads
High Strand-Specificity
QuantSeq-Pool maintains exceptional strand-specificity of >99.9 % and allows to map reads to their corresponding strand on the genome, enabling the discovery and quantification of antisense transcripts and overlapping genes.
Direct Counting for Gene Expression Quantification
Just one fragment per transcript is produced; therefore, no length normalization is required. This allows more accurate determination of gene expression values and renders QuantSeq the best alternative to microarrays and conventional RNA-Seq in gene expression studies.
Fastest Turnaround for Large-scale Projects
The streamlined QuantSeq-Pool workflow uses batch processing of up to 96 samples after the initial protocol step and allows generation of ready-to-sequence NGS libraries in less than 4.5 hours.
Simple Bioinformatics Analysis
Read mapping is simplified by skipping the junction detection. Reads are generated at the transcripts’ most 3′ end where nearly no junctions are located. Data processing can hence be accelerated by using e.g., Bowtie2 instead of TopHat2.
Flexibility of Throughput
The QuantSeq-Pool kit is set up in a 96 well plate format and can generate up to 36,864 triple indexed libraries combining 96 sample-barcodes with 384 unique dual index combinations. The efficiency of the early-pooling workflow and the ability to combine between 8 and 96 samples in one reaction provides full flexibility for projects of any size.
Unique Molecular Identifiers
UMIs are built-in for QuantSeq-Pool and are introduced in the very first step of the protocol. UMIs are 10 nucleotides long and read out at the start of Read 2. Use the UMI information to identify PCR duplicates and eliminate amplification bias.
“We have established QuantSeq-Pool on lysates in our facility and are very happy with its performance. In our hands, QuantSeq-Pool convinced with very low technical variability which boosts the performance for Differential Gene Expression analysis. QuantSeq-Pool now offers us a robust and time-saving procedure that we can scale up to 1,000s of samples for our customer projects.”
Pieter Mestdagh, Principal Scientist, Biogazelle, Belgium
Workflow

The kit uses total RNA as input, hence no prior poly(A) enrichment or rRNA depletion is needed. For balanced read distribution in the sequencing experiment the RNA input
amount for reverse transcription needs to be adjusted and normalized across all
samples that shall be pooled.
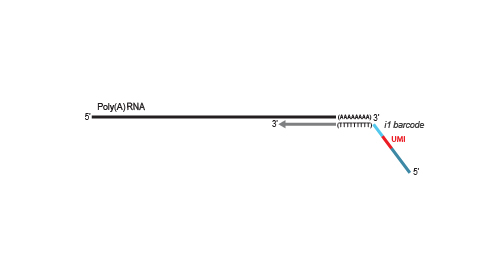
Library generation is initiated by oligo(dT) priming. The primer already contains a partial Illumina-specific Read 2 linker sequence, Unique Molecular Identifiers (UMIs), and an i1 sample-barcode to uniquely label each sample.
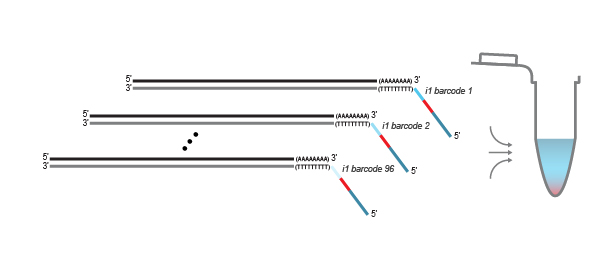
After first strand synthesis, individual samples are combined by pooling. The pool is purified to decrease the volume for subsequent steps. All further reactions are carried out in batch
on the combined samples to save time and effort.
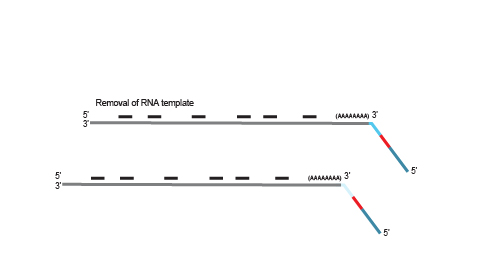
After pooling, the RNA template is removed from all samples simultaneously.
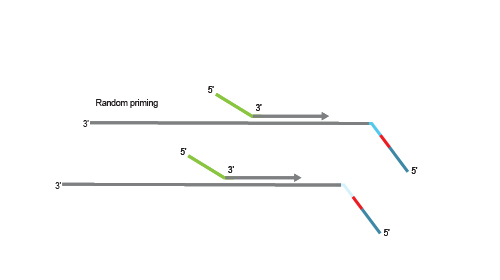
Second strand synthesis is initiated by random priming and a DNA polymerase. The random primer contains the Illumina-specific Read 1 linker sequence.
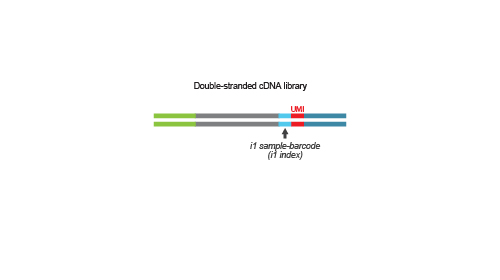
Second strand synthesis is followed by a magnetic bead based purification step.

During the library amplification step sequences required for cluster generation are introduced. Optionally, i5 and i7 indices can be introduced. These additional indices are required for multiplexing more than 96 samples.
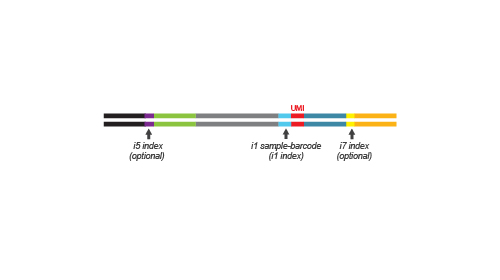
Multiplexing capacities can reach up to 9,216 samples when 96 additional i5 / i7 Unique Dual Indices (UDIs) are used or up to 36,864 samples when 384 UDIs are used.
The final purification elutes ready-to-sequence library pools of up to 96 samples which directly correspond to the final lane pool for a 96-plex sequencing experiment.
NGS reads are generated towards the poly(A) tail and directly correspond to the mRNA sequence. The length distribution of QuantSeq-
Pool libraries supports all sequencing lengths. Read 2 is required to read out the sequence information for the UMI and the i1 sample-barcode. Therefore, at least a limited Read 2 with a read length between 18 and 22 nucleotides is mandatory for sequencing QuantSeq-Pool libraries.
FAQ
Frequently Asked Questions
Access our frequently asked question (FAQ) resources via the buttons below.
Please also check our General Guidelines and FAQ resources!
How do you like the new online FAQ resource? Please share your feedback with us!
Downloads
QuantSeq-Pool Sample-Barcoded 3′ mRNA-Seq Library Prep Kit for Illumina
Safety Data Sheet
If you need more information about our products, please contact us through support@lexogen.com or directly under +43 1 345 1212-41.
Ordering information
| Cat. No. | Product Name |
| 139.96 | QuantSeq-Pool Sample-Barcoded 3’ mRNA-Seq Library Prep Kit for Illumina, 96 preps (For multiplexing of more than 96 samples additional indexing is required. We recommend the Lexogen UDI 12 nt Sets (Cat. No. 101 – 105)) |
Data Analysis
QuantSeq-Pool libraries contain the Read 1 linker sequence in the 5’ part of the second strand synthesis primer, hence NGS reads are generated towards the poly(A) tail. Demultiplexing of i5 / i7 indices can be carried out by the standard Illumina pipeline. Lexogen 12 nt UDI’s i7 and i5 index sequences, as well as i1 sample-barcode index sequences, are available for download at www.lexogen.com. The i1 sample-barcode is located at the beginning of Read 2 and preceded by a 10 nt UMI sequence. Therefore, the i1 barcode is contained within bases at position 11 – 22 of Read 2, depending on the chosen index read out length of 8, 10 or 12 nucleotides. Demultiplexing of i1 sample-barcoded libraries can be performed using Lexogen’s Demultiplexing Tool. All further steps for Trimming, Mapping, UMI processing, and Counting can be integrated in standard data analysis pipelines. Lexogen offers a QuantSeq-Pool data analysis pipeline, which can be found on our GitHub page. For further questions on QuantSeq-Pool data analysis, please contact support@lexogen.com.
Buy from our Webstore
Need a web quote?
You can generate a web quote by Register or Login to your account. In the account settings please fill in your billing and shipping address. Add products to your cart, view cart and click the “Generate Quote” button. A quote in PDF format will be generated and ready to download. You can use this PDF document to place an order by sending it directly to sales@lexogen.com.
Web quoting is not available for countries served by our distributors. Please contact your local distributor for a quote.




