
After extracting RNA from your samples, removing residual DNA, and checking the quality of your RNA preparations, you need to decide if and how to enrich the RNA population of interest. Total RNA is comprised of large amounts of ribosomal RNA (rRNA) which can make up between ~80 – 98 % of all RNA molecules in a sample. For most RNA-Seq applications, the removal of rRNA or the enrichment of polyadenylated transcripts is required to focus the sequencing capacity on the desired parts of the transcriptome and to economize the experiment.
Apart from ribosomal RNA, samples can contain additional abundant transcripts, e.g., globin mRNA in blood samples can account for ~30 – 80 % of all mRNA molecules. Without the removal of abundant transcripts, the majority of reads obtained from an RNA-Seq experiment would be derived from these undesired RNA species taking up valuable sequencing space and limiting the multiplexing capacities within the experiment. Several methods can be used for enrichment of desired RNA, or depletion of undesired RNA which we will discuss in this chapter.
1. Poly(A) Enrichment
The most common pre-processing method used in RNA-Seq is poly(A) selection. Poly(A) selection is used to “fish” polyadenylated RNA species from a total RNA solution. Thereby mature, coding mRNAs are enriched providing the basis for mRNA-Seq workflows. During this procedure, the RNA is first denatured to remove secondary structures and make the poly(A) tails accessible. Oligo(dT) stretches attached to a solid surface, most often to magnetic beads, are then hybridized to the poly(A)-containing RNA molecules. Following hybridization, the supernatant consisting of non-polyadenylated species is removed. The beads are washed prior to elution of the poly(A)-selected RNA. Elution is achieved by elevating the temperature, so that the poly(A) – poly(T) base pairing interaction is resolved, and the selected RNA is eluted from the beads into the solution (Fig. 1).
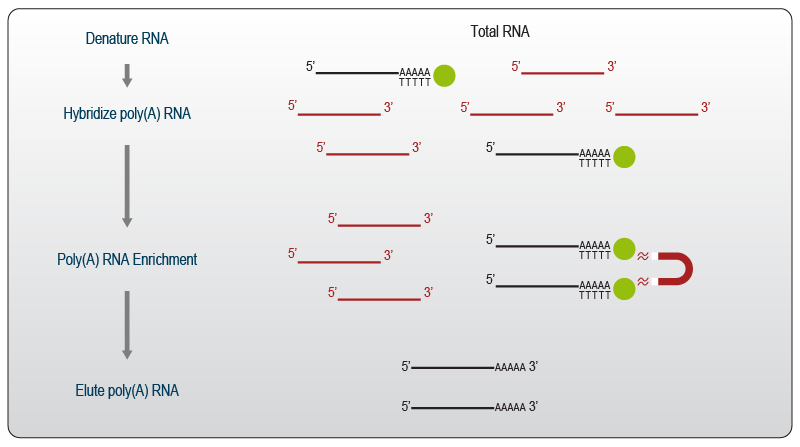
Figure 1 | Schematic overview of poly(A) RNA selection. The workflow is adapted from Lexogen’s Poly(A) Selection Kit.
The selection process excludes the majority of rRNA molecules as these are not polyadenylated. Only ~2 – 5 % of reads are commonly mapped to mitochondrial rRNA, which carries a poly(A) tail and is co-purified.
Poly(A) enrichment is a very cost-efficient and fast pre-processing method that allows selection of mainly protein-coding mRNA. It can be used for all species that possess poly(A)-tailed RNA to remove undesired rRNA and concentrate sequencing reads on mRNA.
However, there are two main drawbacks of poly(A) enrichment:
➊ First, it can only be used for species that possess poly(A)-tailed RNAs. Therefore, it is restricted to eukaryotes and cannot be used for prokaryotes. However, even eukaryotes possess transcripts of interest that lack poly(A) tails. They will be removed together with rRNA during poly(A) selection. These transcripts encompass microRNAs, small nucleolar RNAs (snoRNAs), transfer RNAs (tRNAs), some long non-coding RNAs (lncRNAs), and even protein-coding mRNAs such as histone mRNAs. As a result, researchers interested in those types of RNA or prokaryotic species commonly utilize rRNA depletion instead of poly(A) selection.
➋ Second, poly(A) enrichment requires high quality RNA (RIN / RQN > 8). Degradation leads to breaks within the transcript body and due to the selection of the poly(A) tail, the 3’ ends are enriched while the more 5’ sequences would not be captured, leading to a strong 3′ bias for degraded RNA inputs. Therefore, ribo-depletion or 3’ mRNA-Seq are the methods of choice for working with degraded RNA.
2. Poly(A) Enrichment During Library Preparation
It is also possible to select for polyadenylated transcripts during library preparation by using oligo(dT) priming during reverse transcription. In this case, cDNA is generated primarily starting from the 3’ UTR right at the beginning of the poly(A) tail of mRNAs. This eliminates the requirement for poly(A) selection by the magnetic bead-based approach described above. Therefore, this principle is commonly used in 3’ mRNA-Seq protocols, such as QuantSeq. The complete workflow for 3’-Seq is efficiently shortened and due to the focus on 3’ ends it is also suitable for use with degraded RNA.
It is also possible to generate full-length cDNA by oligo(dT) priming. For these protocols, the reverse transcription reaction is optimized for the generation of long fragments and the 5’ end is routinely enriched by cap-dependent capture methods or template switching.
3. Enzymatic Removal of Abundant Transcripts – Duplex-specific Nuclease (DSN) Treatment
Duplex-specific Nuclease (DSN) is a thermostable nuclease isolated from Kamchatka crab. The enzyme possesses a strong affinity for double-stranded DNA (dsDNA) which is cleaved efficiently while enzyme activity towards single-stranded DNA (ssDNA) is limited. DSN has been used in life science fields for DNA copy number normalization and is not only used in Next-Generation Sequencing (NGS) approaches1,2 but also in forensic analysis of low copy number DNA.
Different DNA copy numbers in an RNA-Seq experiment are the result of two main factors:
➊ The expression level of transcripts in a cell varies between several orders of magnitude: while some transcripts can be present with more than 10,000 copies per cell, others may only be expressed at a very low level with only 1 – 2 copies.
The most abundant transcripts in total RNA samples are rRNA, tRNA, and housekeeping mRNAs. Also, tissue- or sample-specific overabundant transcripts fall into this category.
➋ PCR and amplification bias can lead to preferential amplification of certain molecules while others are under-represented and can thus also be a source of copy number variation.
In RNA-Seq approaches, DSN treatment is used to partially normalize the concentration of cDNAs that reflect the dynamic range of the transcripts. This is achieved by removal of abundant transcripts. DSN treatment is commonly performed after cDNA first and second strand syntheses, however, it is also possible to use DSN after first strand synthesis when the RNA template is not yet removed.
The DSN reaction then uses the hybridization properties of the newly synthesized cDNA molecules (Fig. 2). Following cDNA synthesis, the molecules are denatured by a brief incubation at a high temperature before the reaction is cooled again. Upon decreasing the temperature, the complementary cDNA strands are re-annealed (a process called renaturation). Due to the higher concentration and thus higher chances to interact with the complementary strand, abundant cDNA strands re-anneal faster and more efficiently than lower abundant cDNAs. Therefore, the majority of double-stranded cDNA will be derived from abundant transcripts, while the cDNA derived from the medium and lowly expressed transcripts will stay single-stranded. The double-stranded cDNA fraction is then cleaved by DSN whereby all abundant sequences are removed and the molecules in the pool are normalized to a similar concentration level. Finally, the remaining cDNA molecules are amplified in a PCR reaction to generate sequencing-ready libraries (Fig. 2).
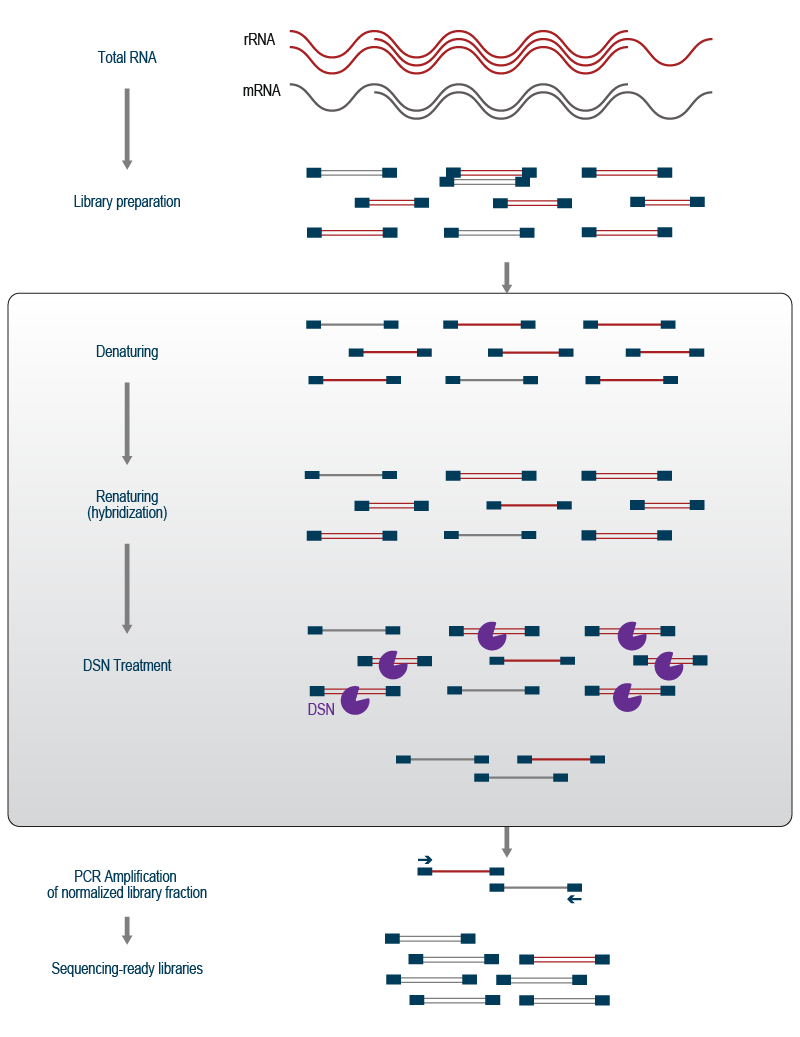
Figure 2 | Enzymatic removal of abundant sequences using Duplex-specific Nuclease (DSN) treatment.
DSN treatment is routinely used in a wide range of applications, but especially for annotation-based approaches as it is quite universal and has the potential to deplete any abundant sequence. It is useful for obtaining transcriptome information from less characterized species for which targeted depletion methods using specific probes are not available, as well as for species that do not possess poly(A) tails and therefore cannot be enriched for mRNA by poly(A) selection 3. Some RNA-Seq kits and universal depletion methods therefore use DSN treatment in their workflows.
The major drawback of this method is that the depletion is unspecific and targeted towards any abundant sequence. If your transcript of interest belongs to the higher-copy number group, it might also be subject to DSN-mediated degradation. In addition, special care should be taken when quantitative information is required to evaluate changes in expression levels. Depending on the input used, DSN treatment may normalize the concentrations in a way that makes it impossible to quantify the changes in expression level correctly.
4. Probe-based Depletion Techniques
In contrast to the unspecific removal of abundant sequences described above, probe-based depletion methods offer the advantage of specifically targeting undesired sequences for removal. This minimizes collateral damage by off-target removal of desired sequences and maintains transcript expression patterns. The downside of this approach is that it requires intricate knowledge of the organism of interest to design the appropriate probe sequences for specific depletion. Probe-based depletion is most commonly used for removal of rRNA transcripts. However, it is also possible to target other abundant sequences for depletion. There are various flavors of probe-based techniques that we will cover in the following section.
Hybridization / capture-based depletion methods use a set of affinity probes that specifically target rRNA sequences. The number and positioning of probes varies depending on the number of species targeted, the complexity of the ribosomal RNA sequences in the target groups, and the compatibility for targeting of degraded RNA. For efficient depletion of rRNA from degraded samples, the density of probes on the targeted sequences needs to be higher as breaks in the target region will impair the hybridization of probes at elevated temperatures. The probes contain affinity tags that allow their capture using magnetic beads with corresponding binding sites. Thus, the number of probes contained in a probe mix is closely related to the binding capacity of the beads used for capture. Increasing the number of probe molecules, e.g., by using a very high frequency of probes or targeting a large group of diverse species can be counter-productive for depletion efficiency as it might overwhelm the capacity of specific binding sites provided by the depletion beads. To ensure optimal results, probes for hybridization / capture methods and depletion beads for commercially available solutions are titrated in the optimal ratio.
In the first step, affinity probes are mixed with total RNA and denatured, thereby facilitating access of probes to highly structured target sequences. Hybridization is performed at an elevated temperature to ensure specific binding and to minimize undesired off-target depletion. Depletion beads are used to remove probes that are hybridized to ribosomal RNA from the solution. A final purification step removes all reaction components and recovers pure depleted RNA for downstream applications (Fig. 3).
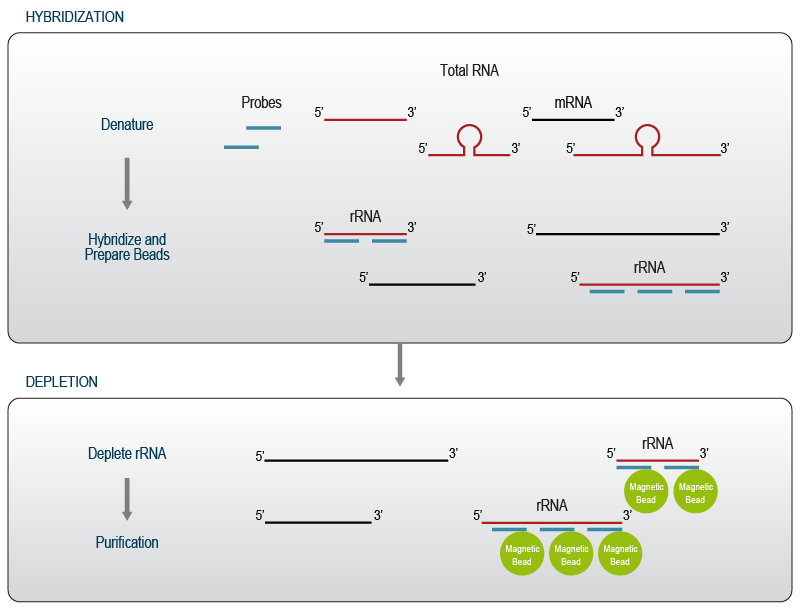
Figure 3 | Depletion of rRNA using specific probes for hybridization / capture. The workflow is adapted from Lexogen’s RiboCop Depletion Kits.
Hybridization / capture methods do not rely on enzymatic reactions and therefore these methods leave full-length transcripts intact for downstream processing. They are particularly useful for challenging applications, such as RiboSeq 4, and minimize unspecific RNA degradation.
Here at Lexogen, we aim to provide complete workflow solutions by integrating selection and depletion kits with our innovative library preps. To learn more about our solutions, visit our blog article.
RNase H is an endoribonuclease that specifically cleaves the RNA molecule within an RNA:DNA duplex while keeping the DNA molecule intact. It is often used in probe-based depletion protocols and falls into the class of specific, enzyme-mediated depletion. This depletion method uses specific DNA probes that hybridize to the target molecule, commonly rRNA and / or globin mRNA. The density of the probes used in the reaction can vary. It is even possible to cover the complete transcript multiple times by using partially overlapping, or so-called “tiling” probes. For rRNA depletion using RNase H, the DNA probes are hybridized to the rRNA at elevated temperatures. After the probes are bound, the reaction is incubated with RNase H which specifically degrades the rRNA.
Depending on the workflow, it is also possible to use a thermostable variant of RNase H (Hybridase™) and incubate the reaction at a temperature of 65 °C or above. This allows increased stringency of the depletion reaction by minimizing nonspecific hybridization at lower temperatures.
Following RNase H treatment, the reaction is incubated with DNase to remove the oligonucleotide probes before the reaction is purified to remove all enzymes and reaction components and elute the now depleted RNA sample (Fig. 4).
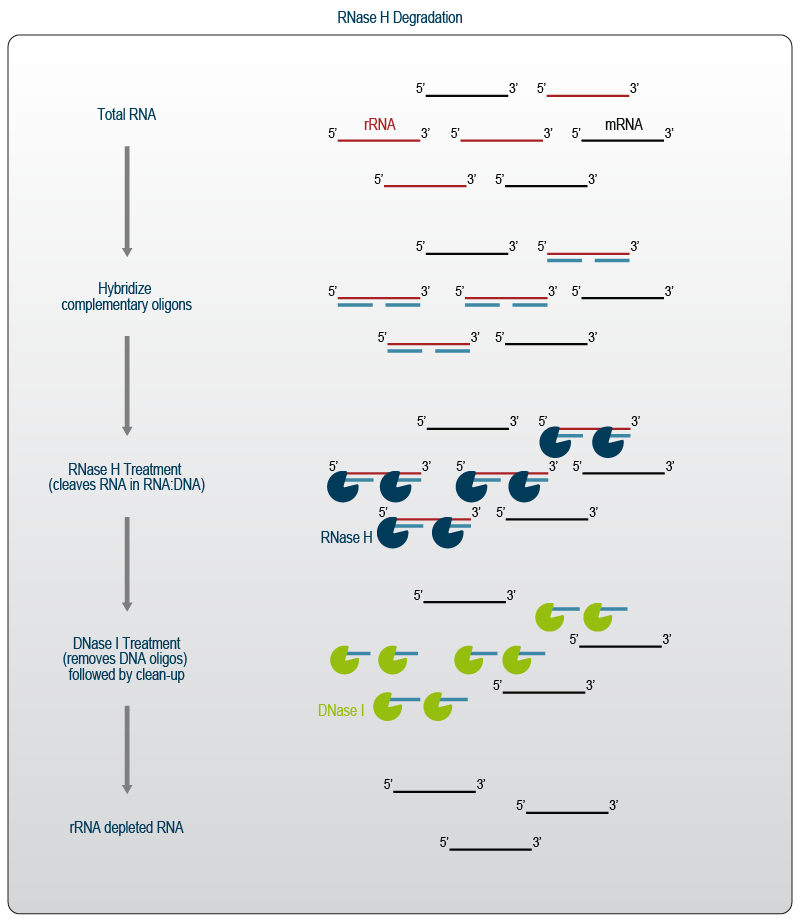
Figure 4 | RNase H-mediated depletion of rRNA using specific probes.
RNase H-based degradation is widely used as the reaction components and oligos are cheap and their number can be increased to cover many species. However, the nature of the method being based on degradation poses the risk of unspecific removal of precious transcripts and also makes it unsuitable for certain applications. For example, RNA-Seq workflows that rely on the addition of DNA-adapters prior to depletion should not undergo RNase H / DNase I treatment, as this will degrade the RNA-DNA fusion molecule as well. Recent findings suggest that RNase H-based approaches may not be suitable for challenging applications such as RiboSeq 4.
CRISPR (clustered regularly interspaced short palindromic repeats) and Cas (CRISPR associated) nucleases, such as Cas9, gained wide popularity in recent years as the “gene scissors”. The system has revolutionized genome editing and has a wide range of applications ranging from providing essential molecular biology research tools to solutions for personalized medicine. The groundbreaking discovery by Emmanuelle Charpentier and Jennifer Doudna 5, 6 was awarded the Nobel Prize in chemistry in 2020.
The natural CRISPR-Cas system is functional in bacterial adaptive immunity and removes incoming phage DNA without harm to the bacterial genome. The mechanism involves specific cleavage of the foreign DNA by Cas nuclease. Specific guide RNAs which are transcribed from the CRISPR loci bind to the Cas nuclease and guide the enzyme to the complementary target DNA which is then neutralized by Cas-mediated cleavage.
The system can be exploited by providing specialized guide RNAs to target any desired sequence. In RNA-Seq approaches, CRISPR-Cas9 is used together with guide RNAs targeting rRNA sequences or other abundant sequences and can conveniently be used after the library preparation is completed.
The ready-to-sequence libraries are incubated with Cas9 nuclease that has been pre-complexed with the specific rRNA-guides (Fig. 5). All libraries containing any of the targeted sequences are then cleaved within the pool of molecules. The incubation is followed by a clean-up step to remove the cleaved fragments which are shorter than the remaining non-targeted libraries and are no longer tagged with both sequencing adapters. As this process removes the majority of library fragments, the remaining molecules are re-amplified by an additional round of PCR.
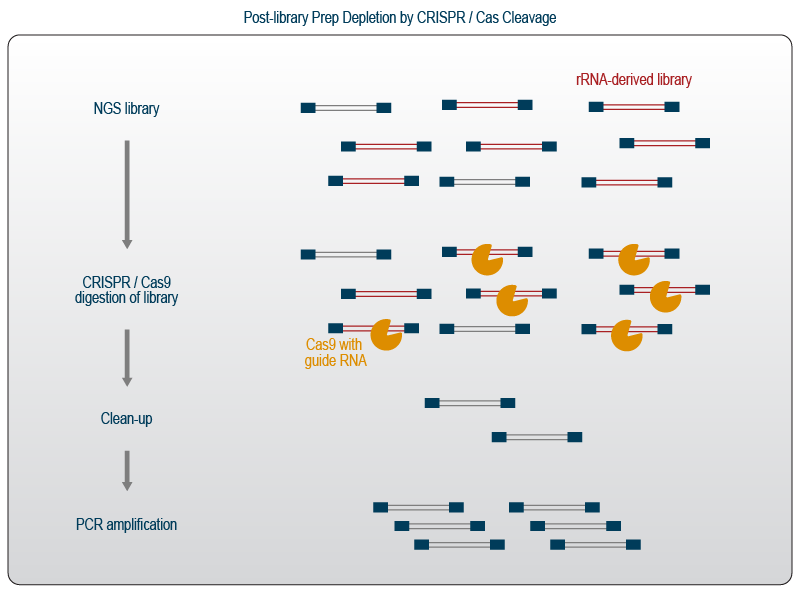
Figure 5 | CRISPR-Cas9-directed post-library prep depletion of rRNA and abundant sequences.
Post-library prep depletion is advantageous when working with samples that do not allow depletion prior to library preparation, e.g., when working with very low input amounts or addressing single cells without oligo(dT) priming. It also allows the researcher to deplete final lane mixes, which saves on depletion cost. On the downside, guide RNA design is quite complex, which may limit the customization potential for non-expert users. Further, libraries for sequencing need to undergo two rounds of PCR which may increase amplification bias. To preserve the guides, storage at -80 °C is advised.
5. In-prep Depletion using Target-specific Blockers
In-prep depletion solutions use sequence-specific blocker oligos that prevent the generation of ready-to-sequence libraries from undesired sequences. Blocker oligos can be used during the reverse transcription step to prevent first strand cDNA generation from these sequences, or they can be added during second strand synthesis to prevent generation of the complementary cDNA strand. To prevent polymerization and strand-displacement activity of the enzymes catalyzing these reactions, blocker oligos are heavily modified to ensure their inertness.
The use of blocker oligos during second strand synthesis allows blocking of abundant sequences also in 3’ mRNA-Seq methods. This increases complexity from samples containing abundant mRNAs and elevates the gene detection capacity without the need to increase sequencing depth. One commonly used blocker oligo mix for 3’ mRNA-Seq allows removal of reads derived from globin mRNA.
6. Exclusion of Abundant Transcripts during sRNA Extraction
Small RNAs (sRNAs) are essential regulators of gene expression and involved in regulatory pathways, such as cancer, inflammation, and development. Isolation with column-based sRNA extraction kits does not confer any specificity since all RNAs below a common threshold of 200 nucleotides are purified. As a result, the majority of sRNA-Seq reads usually correspond to non-functional RNA degradation fragments mostly derived from rRNA and tRNA. To concentrate the sequencing reads on sRNAs, additional selection methods are used, such as size selection by gel extraction or chemical treatment. These methods are extremely laborious, time intensive, and are associated with sample losses. A fast and convenient extraction method, called TraPR, was recently developed by a group of sRNA researchers at ETH Zurich7. This method uses a convenient column-based centrifugation step prior to RNA extraction that enriches sRNAs in their functional protein-bound form and removes any free RNA and DNA from the sample. Subsequent RNA extraction then results in a pure fraction of functional sRNA without undesired rRNA fragments, which ultimately saves sequencing space and allows multiplexing of more samples. To find out more about the technology, please see our short RNA Expertise video on TraPR.
Are you ready to become an RNA Expert?
Sign up and gain access to helpful checklists in PDF format that can assist you in your experiments. In addition, you’ll have the opportunity to download the RNA LEXICON E-BOOK in PDF format as well.
Literature:
1 Shagina, I., Bogdanova, E., Mamedov, I.Z., Lebedev, Y., Lukyanov, S., and Shagin, D. (2010) Normalization of genomic DNA using duplex-specific nuclease. Biotechniques. 48:455-9. DOI: 10.2144/000113422.
2 Bogdanova, E.A., Shagin, D.A., and Lukyanov, S.A. (2008) Normalization of full-length enriched cDNA. Mol Biosyst. 4:205-212. DOI: 10.1039/b715110c.
3 Yi, H., Cho, Y.J., Won, S., Lee, J.E., Jin Yu, H., Kim, S., Schroth, G.P., Luo, S., and Chun, J. (2011) Duplex-specific nuclease efficiently removes rRNA for prokaryotic RNA-seq. Nucleic Acids Res. 39:e140. DOI: 10.1093/nar/gkr617.
4 Zinshteyn, B., Wangen, J. R., Hua, B., and Green, R. (2020) Nuclease-mediated depletion biases in ribosome footprint profiling libraries. RNA 26: 1481-1488 DOI: 10.1261/rna.075523.120.
5 Deltcheva, E., Chylinski, K., Sharma, C. et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. 2011 Nature 471, 602–607 DOI: 10.1038/nature09886.
6 Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J.A., and Charpentier, E.(2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816-821. DOI: 10.1126/science.1225829.
7 Grentzinger T., Oberlin, S., Schott, G., et. al. (2020) A universal method for the rapid isolation of all known classes of functional small RNAs. Nucleic Acids Res., DOI: 10.1093/nar/gkaa472.




