Next-Generation Sequencing (NGS) is the gold-standard for genomic and transcriptomic research in life science fields . While it shares certain core similarities with methods such as Sanger sequencing, the massive level of throughput made possible by NGS sets it worlds apart and has revolutionized the way scientists work.

RNA Sequencing (RNA-Seq) is at the cutting edge of NGS capabilities. In its simplest form, RNA-Seq allows us to determine RNA molecules in a sample at the moment of sampling. The transcriptome is a highly dynamic cellular feature that opens up a world of discovery potential. Changes in response to drugs, various states of disease, post-transcriptional modifications, and alternatively spliced transcripts are just some examples of discoveries made possible by RNA-Seq.
In short, RNA-Seq allows us to assess the whole transcriptome at unprecedented levels of sensitivity and generate a snapshot of the transcriptome under conditions and points in time specific to the topic being studied. Though this serves as a perfectly suitable and brief summary of the broader field of RNA-Seq, there are many “flavors” of RNA-Seq to try.
Short-read sequencing is performed commonly on RNA-Seq library preps. Sequencing is typically done on shorter inserts generated from the sample material between 50 – 500 bp in length and then re-assembled or counted in downstream data analysis. Long-read sequencing is frequently used as a DNA-Seq method as well as an RNA-Seq method and can determine the sequence of the sample at hand at a high capacity, e.g., between 10,000 – 100,000 base pairs at a time. For RNA Sequencing, combining long-read and short-read sequencing is especially useful to determine the transcriptomes of unknown or under annotated species. Long-read sequencing can determine the exact transcript sequence and provide a scaffold on which the short reads can be assembled similar to puzzle pieces.

Bulk RNA-Seq measures the gene expression of a set of samples without differentiating between the cell types within the sample. This method provides a broad overview of gene expression in a set of samples. The range of RNA-Seq applications is extremely broad, and the boundaries are being pushed with every passing day (see the List below for example applications). With such a wide range of applications, it is no surprise that RNA-Seq is a useful, powerful tool that offers quantitative, qualitative, and time-resolved data on set of experimental samples.
Here at Lexogen we are focused on the short-read RNA sequencing workflow. While there are exceptions to this, the general outline of an experimental procedure is as follows:
➊ RNA is isolated from the sample and contaminating DNA is removed, e.g., with DNase treatment.
➋ If needed for the method, the RNA is pre-treated, i.e., the mRNA is enriched by poly(A) selection or rRNA depletion.
Very commonly a 3’ mRNA library generation method is used which enriches for poly(A) tail-containing material, as this region is rich in features and enables sound differential gene expression studies without over-working the samples ahead of time.
➌ RNA can then be fragmented. If using fragmentation-free protocol, this point can be ignored.
➍ Reverse transcription is performed using the RNA sample and library generation primers as chosen.
➎ A second strand synthesis is then performed by randomly priming along the first cDNA strand.
➏ At this point the libraries are ready for end repair and the ligation of any products occurs on either double stranded cDNA.
➐ PCR is finally performed to add indices to the library, and / or amplify the material for library quality control.
The individual steps to prepare NGS libraries and the molecular principles underly these reactions are described in more detail in Chapter 7: RNA-Seq Library Preparation – Molecular Biology Basics.
Lexogen library preps for mRNA-Seq and whole transcriptome sequencing use a slightly different approach. Partial adapters are introduced already in the first step, e.g., during reverse transcription. Thereby, workflows are streamlined and efficiently shortened by removing various processing steps. This saves valuable time and allows to complete the library preparation workflow within a few hours. Figure 1 illustrates the schematic workflow for two different RNA-Seq library preparation methods.
RNA-Seq can be used for quite a variety of applications, is highly customizable by the combination of different methods, and offers sheer endless possibilities for modifications to fit the individual needs of a research project. Planning your experiment before taking up the pipets is the key to success and ensures high quality results to foster your ideas and hypotheses.
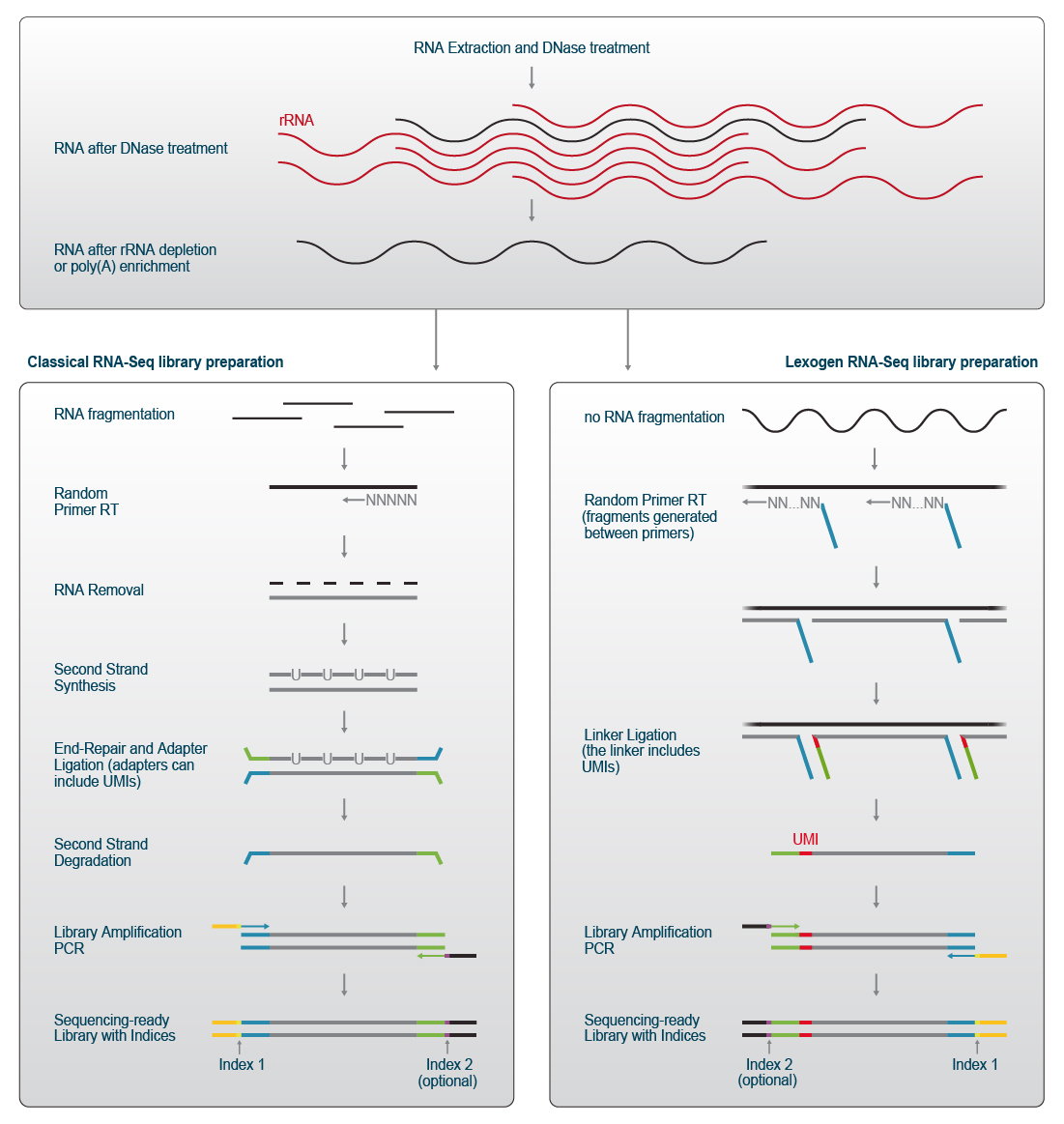
Figure 1 | RNA-Seq Library Preparation Workflows for Illumina short-read sequencing. RNA is extracted from cells, tissues, bioliquids or other samples and treated with DNase to remove genomic DNA prior to depletion of ribosomal RNAs or poly(A) RNA enrichment.
Are you ready to become an RNA Expert?
Sign up and gain access to helpful checklists in PDF format that can assist you in your experiments. In addition, you’ll have the opportunity to download the RNA LEXICON E-BOOK in PDF format as well.




