After you have successfully extracted the RNA from your sample, controlled the quality of your preparation, and removed residual gDNA (if needed), it is time to prepare your RNA-Seq libraries. Depending on the library method you have chosen and the RNA fraction you are interested in, you may need to pre-select for your RNA fraction of choice. For more details on this, check out our chapter on RNA Enrichment and Depletion.
While the individual reaction steps in an RNA-Seq workflow can vary depending on the library preparation method used, the molecular principles underlying these reaction steps remain the same. The following reactions are commonly used in RNA-Seq library preparation: Reverse Transcription, Second Strand Synthesis, End Repair, Ligation, and PCR Amplification. In the following chapter, we will go over these steps and shed light on the molecular basis of these reactions.

1. Reverse Transcription / First Strand Synthesis
First strand synthesis refers to the generation of a complementary DNA molecule from an RNA template by an enzyme called Reverse Transcriptase. Reverse transcriptases are a viral RNA-dependent DNA-polymerases that transcribe RNA template molecules into copy DNA (cDNA). The discovery of reverse transcriptases in the 1970s1,2 has revolutionized molecular biology by short-circuiting Francis Cricks’ original dogma of molecular biology which described a unilateral flow of genetic information from DNA → RNA → Protein (Figure 1). This discovery was awarded with a Nobel Prize in 1975.
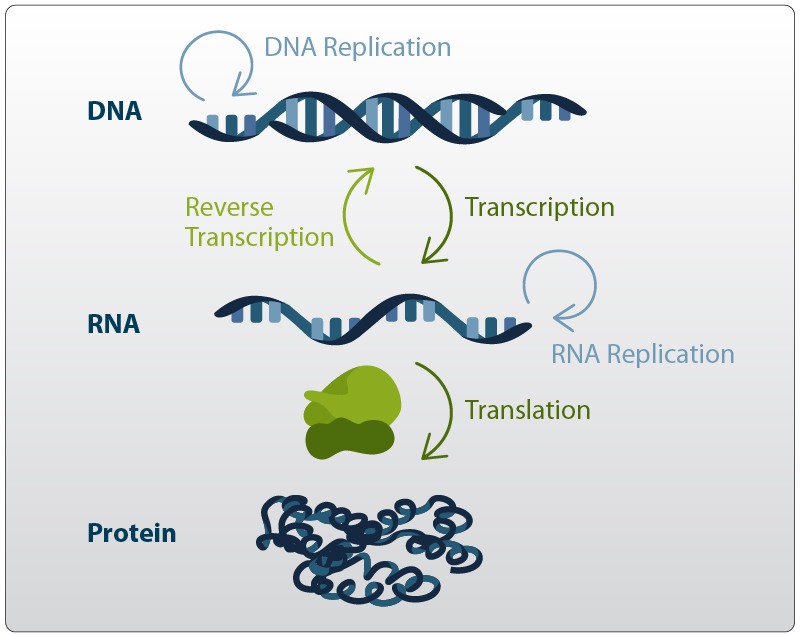
Figure 1 | Central Dogma of Molecular Biology.
The central dogma of molecular biology describes the flow of genetic information within biological systems. DNA can either be replicated by DNA Polymerase (DNA → DNA) or is transcribed into RNA by RNA Polymerases (DNA → RNA). RNA is a messenger molecule that can be translated into proteins by Ribosomes (RNA → protein). Reverse transcription describes the conversion of RNA into DNA (RNA → DNA) by reverse transcriptase and is commonly used by viruses. RNA replication generates RNA from RNA.
Today, reverse transcription is at the core of all RNA-Seq experiments and as such remains one of the fundamental reactions in state-of-the-art molecular biology applications.
A reverse transcription reaction in an RNA-Seq workflow requires three sub-steps: primer annealing, cDNA synthesis (Fig. 2), and enzyme inactivation.
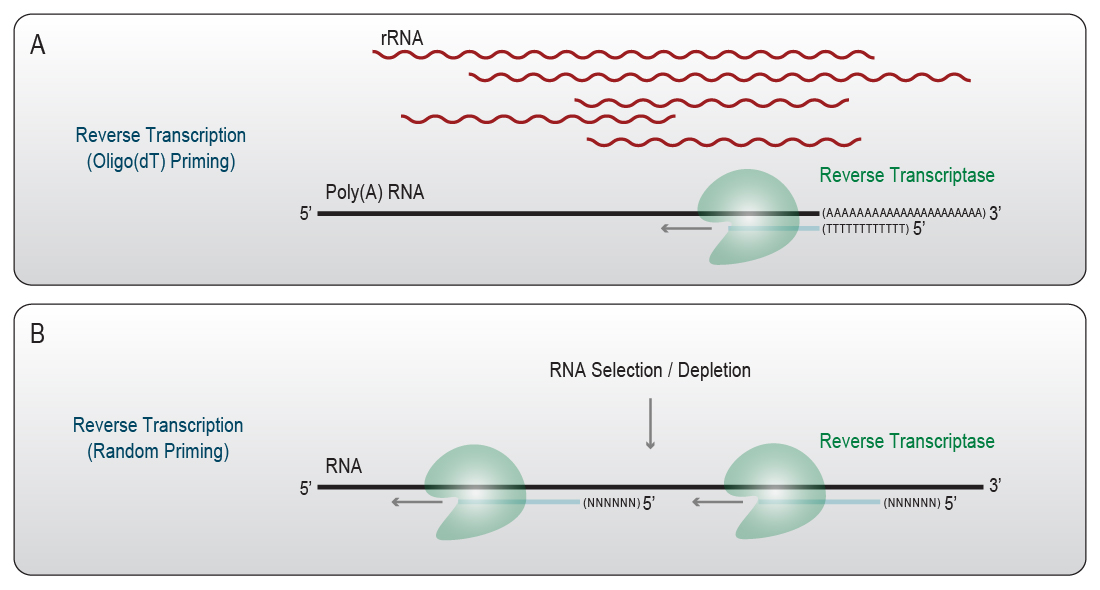
Figure 2 | Reverse Transcription generates cDNA from RNA template molecules. A) Oligo(dT)-primed reverse transcription does not require rRNA depletion or mRNA selection due to the primer annealing specifically to the 3’ poly(A) tail of mRNAs. B) Random-primed reverse transcription uses pre-selected or depleted input RNA. Random primers hybridize along the RNA template. Reverse transcriptase elongates the primers and generates a complementary DNA copy.
2. RNA Template Removal and Second Strand Synthesis
After the first DNA strand is generated, many RNA-Seq workflows require the generation of a double stranded DNA molecule. The product of the reverse transcription reaction is a cDNA single strand that is paired with the initial RNA template strand. In order to generate the second DNA strand, the RNA first needs to be removed. There are different ways to achieve RNA removal. Most commonly, the product is heated in a buffer that is formulated to specifically hydrolyze RNA while the DNA strand stays intact.
Subsequently, random primers are annealed to the now accessible cDNA first strand and the second strand is generated by incorporation of complementary nucleotides using a DNA-dependent DNA Polymerase. For random primed second strand synthesis, DNA polymerases that work at lower temperatures are used, while their thermostable counterparts are mostly used in PCR amplification and for targeted RNA-Seq approaches.
RNA template removal is required to ensure that short random primers used to initiate second strand synthesis gain access to the cDNA first strand. In case a targeted sequencing approach is used, RNA removal can be omitted. Targeted primers are commonly designed with a high annealing temperature (>60 °C) to avoid unspecific priming to non-target sequences. Concomitantly, the reaction is carried out at a much higher temperature than random primed second strand synthesis. This elevated temperature of 60 – 72 °C weakens the RNA-DNA interactions sufficiently for the targeted primer to anneal to the cDNA first strand. The RNA template is unwound and the complementary DNA second strand is generated.
Most DNA-polymerases used for second strand synthesis possess proofreading activity and thus the error rates in this step are much lower than during reverse transcription.
The properties of natural reverse transcriptases in combination with a process called nick translation for second strand synthesis were already exploited in the 1980s for efficient generation of cDNA libraries3.
3. End Repair
First and Second Strand Synthesis generate partially double stranded DNA with single-stranded ends. A process termed end repair is therefore often used to prepare the double-stranded DNA library for the adapter ligation step. During the end repair reaction, partial single strands on the 5’ end of the fragment are filled in by a polymerase to generate a double strand using the protruding end as a template. Single-stranded 3’ overhangs on the other end of the DNA fragment are removed using a 3’ → 5’ exonuclease. This process creates blunt ends on both sides of the fragment.
Depending on the adapter ligation strategy, a single adenine can be added at the 3’ end of each strand in a process referred to as A-tailing. These A-tailed fragments are subsequently ligated to adapters with a single 5’ T-overhang in the subsequent ligation step (Fig. 3).
As the efficiency of blunt end ligation is usually lower than ligation with overhangs (even if it is just one nucleotide), end repair including A-tailing is a common theme in library preparation for Next Generation Sequencing.
As DNA ligases require molecules with a 5’ phosphate (5’-P) and a 3’ hydroxyl group (3’-OH) as substrates, these functional groups are also generated during the end repair process. This is achieved either by using enzymes that generate such end products or by enzymatic activities that transfer these functional groups, e.g., Polynucleotidekinase (PNK) can transfer phosphate groups to the 5’ end of RNA.
There are also other variations of “end repair” used in RNA-Seq library preparation. The common theme is that overhangs are removed or filled in. This is mostly done on DNA, but RNA can also undergo end repair.
4. Ligation
We already outlined a few basic principles of ligation in the previous section, here we will go into a few more details. Ligation in molecular biology refers to the enzymatic process of joining two nucleic acid molecules by attaching the 3’-OH group of the first molecule to the 5’-P group of the second molecule. Ligases were discovered by multiple labs4 in the 1960s and are also considered as one of the major breakthroughs in molecular biology, enabling molecular cloning of recombinant DNA molecules and NGS library generation.
As mentioned above, ligation occurs in various modes.
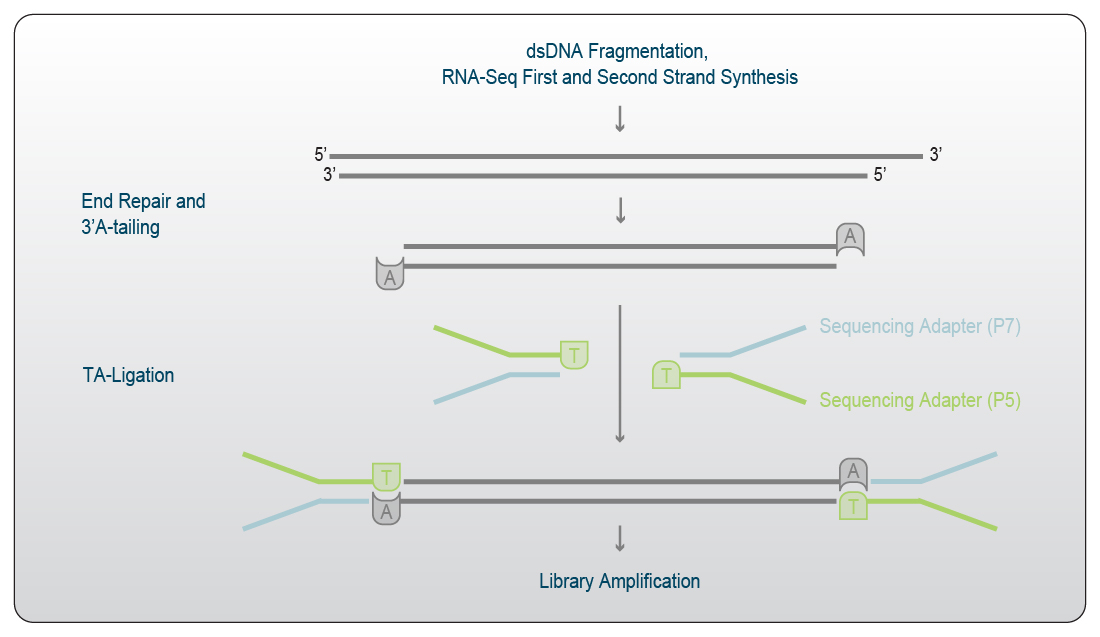
Figure 3 | End Repair, A-tailing and TA ligation. After first and second strand synthesis, single stranded overhangs are removed and the 3’ ends are adenylated. Pre-annealed partially double-stranded sequencing adapters with 3’ T-overhangs are ligated to the A-tailed inserts. Sequencing adapters consist of a P7 and a P5 linker sequence.
5. PCR Amplification
PCR or polymerase chain reaction is a widely used method to generate millions of DNA copies from as low as a single molecule. The description of the PCR reaction5 and its first application in the 1980s is mainly attributed to Kary Mullis who received the 1993 Nobel Prize in Chemistry for his discoveries. The extreme sensitivity and versatility of PCR led to its numerous applications in various fields of science ranging from molecular biology research to medical diagnostics, to infectious disease detection (e.g., SARS-CoV-2 diagnostics), even down to paternity testing and criminal forensics, and has helped to unravel the genome of Neanderthals.

PCR cyclers for running three different PCR reactions in one machine.
Thus, polymerase chain reaction is an integral part of cutting-edge science and used in many state-of-the-art techniques, including high-throughput Next Generation Sequencing. PCR is the final step in most NGS library preparation workflows. During this step, the libraries are amplified for quality control. In case partial adapters were introduced in the library generation step, the adapter sequences are completed and indices are introduced.
PCR uses a thermostable polymerase to amplify a DNA template by repeating three steps in multiple cycles: denaturation, primer annealing, and elongation.
Literature:
1 Temin HM, Mizutani S (1970) RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature 226(5252):1211–1213
2 Baltimore D (1970) RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature 226(5252):1209–1211
3 Gubler U, Hoffman BJ (1983) A simple and very efficient method for generating cDNA libraries. Gene 25(2-3):263-269
4 Lehman IR. DNA ligase: structure, mechanism, and function. Science. 1974 Nov 29;186(4166):790-7. doi: 10.1126/science.186.4166.790. PMID: 4377758
5 Mullis, K.F.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. (1986). “Specific enzymatic amplification of DNA in vitro: The polymerase chain reaction”. Cold Spring Harbor Symposia on Quantitative Biology. 51: 263–273. doi:10.1101/sqb.1986.051.01.032
Are you ready to become an RNA Expert?
Sign up and gain access to helpful checklists in PDF format that can assist you in your experiments. In addition, you’ll have the opportunity to download the RNA LEXICON E-BOOK in PDF format as well.




