
GET THE BEST SEQUENCING RESULTS
with all-in-one library preparation kits
→ Our new CORALL RNA-Seq V2 Library Prep Kits
Do you have any questions?
Find the ideal kit for your application:
CORALL Total RNA-Seq V2
Total RNA-Seq provides the optimal coverage for samples of varying quantity and quality and is perfectly suited for analyzing FFPE and Biobank samples, long non-coding RNA or perform bacterial transcriptomics after ribosomal RNA depletion.
Get the best sequencing data with CORALL Total RNA-Seq V2. The kit enables fast and cost-efficient generation of stranded, UMI labelled, and unique dual indexed libraries with adjustable insert size. Bundles are available including rRNA depletion for human / mouse / rat ribosomal transcripts and globin mRNA for a convenient complete workflow solution.
Performance
Exceptional Gene Detection Across a Wide Range of Input Amounts
CORALL Total RNA-Seq V2 delivers excellent gene discovery rates across a wide range of RNA input amounts for long and medium library lengths (Fig. 1).
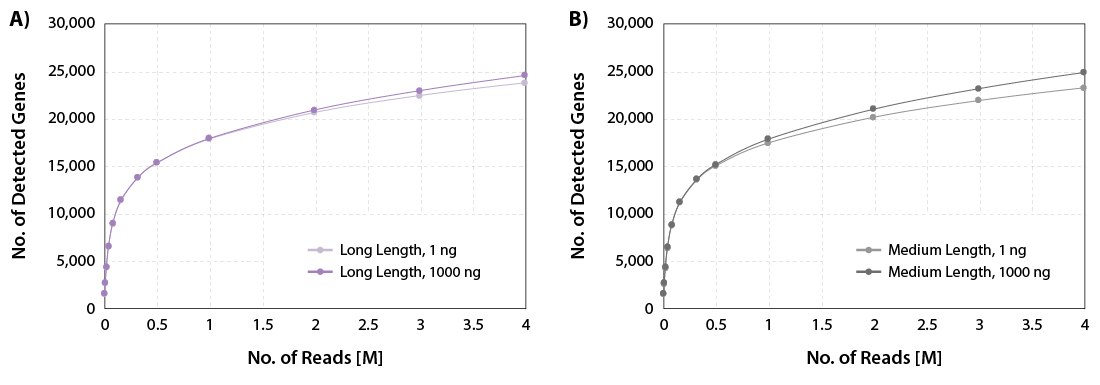
Figure 1 | Gene discovery rates. A) Gene discovery rates for libraries with long length (average size 550 bp) and B) medium length (average size 350 bp). 1 ng, and 1000 ng Universal Human Reference RNA (UHRR) were used as input for rRNA depletion with RiboCop and library preparation using CORALL Total RNA-Seq V2. Libraries were sequenced on Illumina® NextSeq500 (2×150 bp). The number of detected genes is plotted against the number of reads mapping uniquely to exons (calculated with featureCounts).
Excellent Reproducibility and Sensitivity
Correlation analysis of CORALL total RNA-Seq libraries from 1 ng total RNA input prior to rRNA depletion with RiboCop reveals excellent reproducibility for libraries of all sizes (Fig. 2).
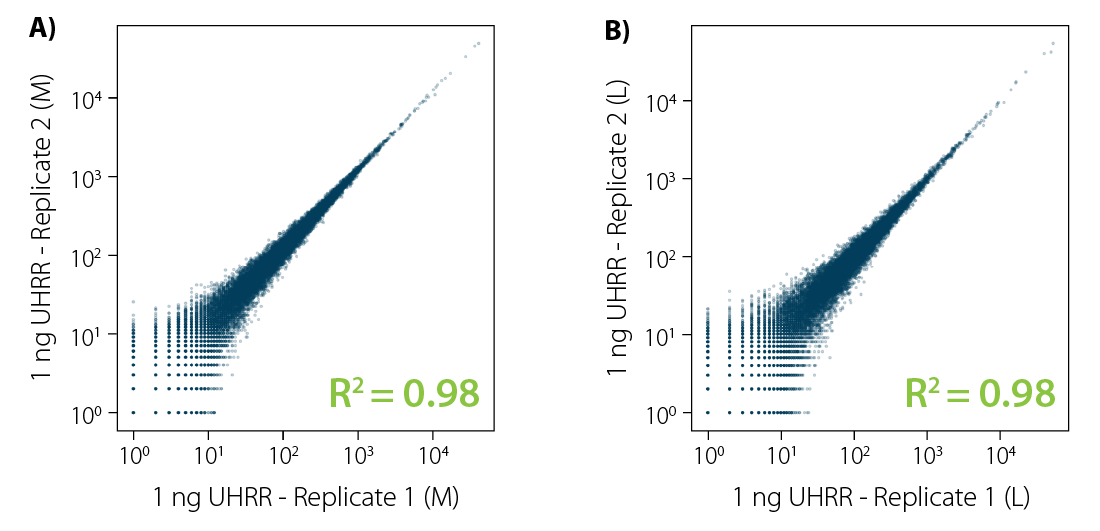
Figure 2 | Excellent reproducibility between replicates for low input RNA. Correlation analysis between replicates for CORALL Total RNA-Seq with 1 ng UHRR input prior to rRNA depletion with RiboCop. Correlation plots for libraries with A) medium length (average size ~350 bp) and B) long length (average size ~550 bp). Libraries were sequenced on Illumina® NextSeq500 (2×150 bp).
Benchmarked Performance
CORALL Total RNA-Seq gene detection matches the performance of renown competitor kits (Fig. 3).
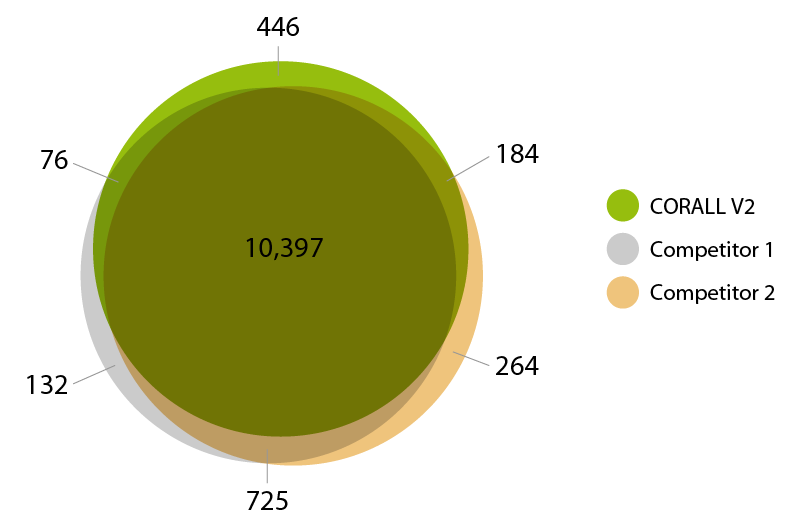
Figure 3 | Gene detection overlap. The Venn diagram illustrates the overlap of detected genes between CORALL RNA-Seq V2 and two competitor kits at normalized expression levels of > 10 CPM.
Transcript Coverage
CORALL RNA-Seq V2 generates transcriptome-wide smooth, uniform read coverage, with exceptional 5’ coverage (Fig. 4). Gene body coverage is comparable to competitor protocols.
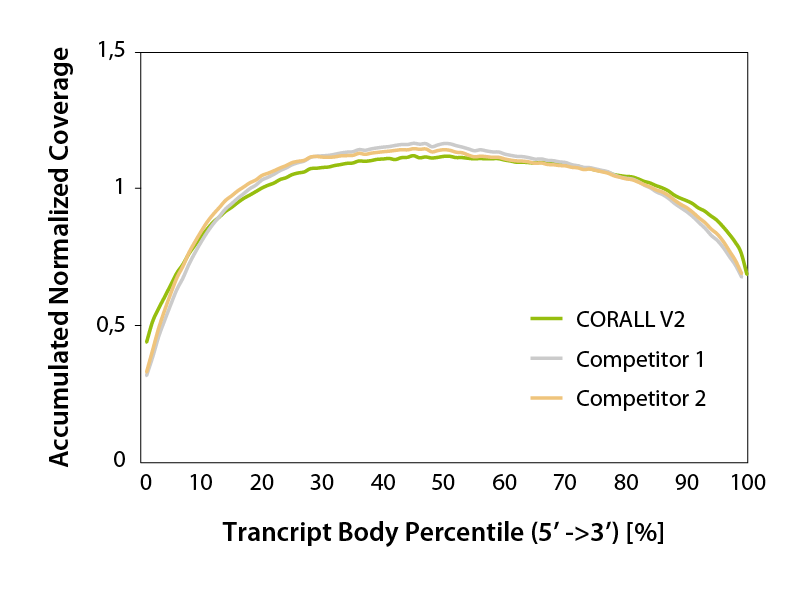
Figure 4 | Accumulated transcript body coverage (whole transcriptome). Coverage across all transcripts was generated using the geneBody_coverage.py tool provided by RSeQC (transcripts length normalized to 100 %).
Performance on FFPE Samples
CORALL RNA-Seq V2 is suitable for processing degraded and compromised samples, including FFPE material. Gene discovery rates are highly comparable between fresh frozen and FFPE matched tissue samples from human liver (Biochain, R8234149-FP). Qualitative analysis shows a large overlap of detected genes at normalized expression levels >5 CPM between FFPE and fresh frozen samples demonstrating consistent and robust performance of CORALL RNA-Seq V2 even for low quality / FFPE RNA samples (Fig. 5).
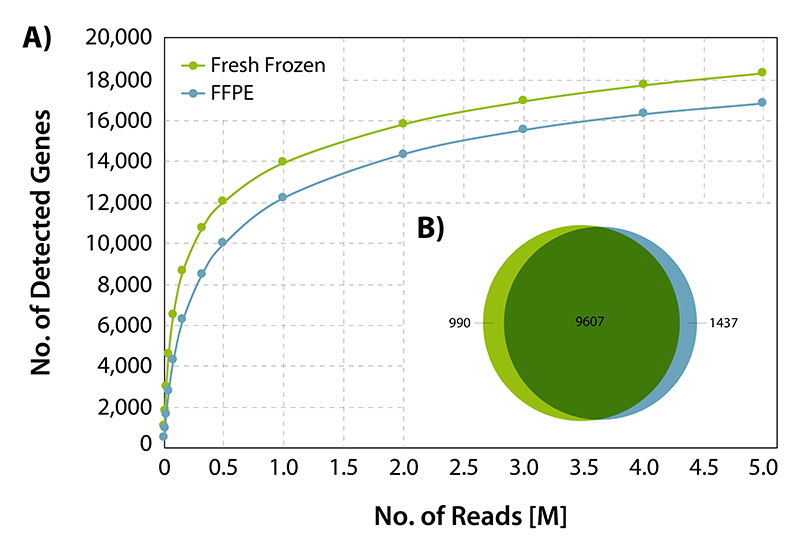
Figure 5 | Gene discovery rates for fresh frozen and FFPE derived RNA. 25 ng of fresh frozen or FFPE fixed human liver RNA were treated with DNase I and depleted for rRNA with RiboCop HMR V2. Libraries were prepared according to user guide, sequenced on NextSeq 500 (1x 50 bp) and analyzed. A) Gene detection rates for fresh frozen or FFPE fixed human liver matched tissue RNA. The number of detected genes is plotted against the total number of reads mapping uniquely to exons. B) Overlap of detected genes from fresh frozen and FFPE input RNA at normalized expression levels >5 CPM (for uniquely mapping reads).
Performance on Blood Samples
Total RNA from mammalian blood can be obtained by minimally invasive sampling and is therefore the most commonly used sample to study diseases in cohorts or patients. However, whole blood RNA is comprised of highly abundant undesired RNA species, such as ribosomal RNA (rRNA), accounting for ~80 – 90 % of total RNA, and globin mRNA, representing 30 – 80 % of all mRNAs.
CORALL Total RNA-Seq V2 combined with RiboCop for Human/Mouse/Rat plus Globin for simultaneous removal of rRNA and globin mRNA provides a highly convenient workflow for processing whole blood samples. Depletion of rRNA and globin mRNA frees up sequencing space for RNAs of interest (Fig. 6) and leads to a significant increase in gene detection (Fig 7 A and B).
This workflow allows to study coding and non-coding RNAs of the blood, including mRNA, lncRNAs, snoRNAs, and snRNAs. For convenience, bundles of CORALL Total RNA-Seq V2 with RiboCop for Human/Mouse/Rat plus Globin are available (Cat. No. 185 and 186).

Figure 6 | CORALL Total RNA-Seq V2 with RiboCop rRNA Depletion for Human/Mouse/ Rat plus Globin efficiently removes rRNA and globin mRNA from human whole blood RNA. RNA was extracted from human donor whole blood. 5 ng whole blood RNA were depleted with RiboCop HMR V2 or HMR+Globin. Libraries were prepared with the CORALL Total RNA-Seq V2 Library Prep and sequenced on NextSeq500 (1×75 bp). Reads were mapped against the human reference genome (GRCh38.95) and reads mapping to rRNA (blue) and globin (purple) were calculated.
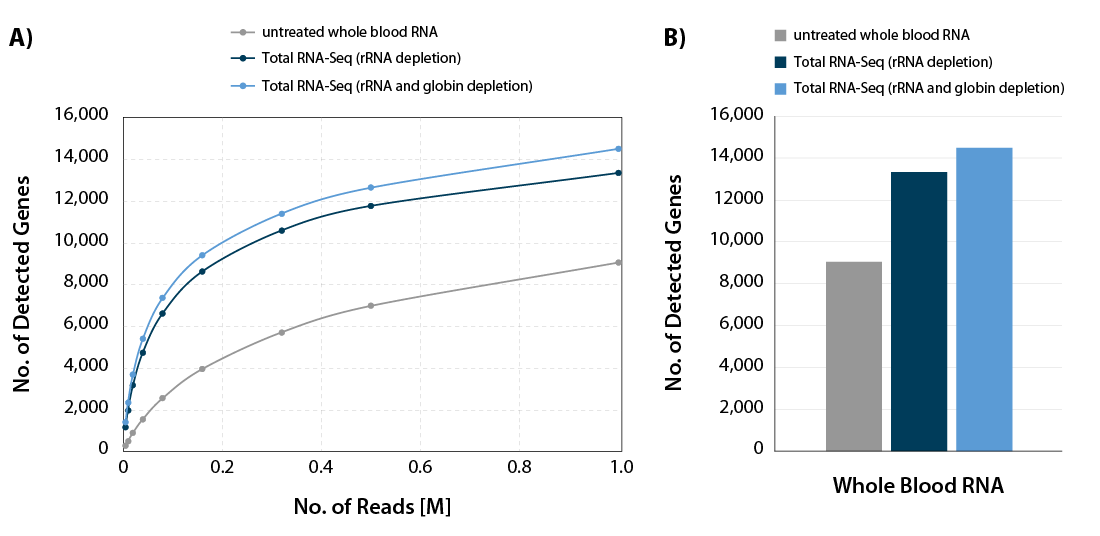
Figure 7 | Increased gene detection for CORALL Total RNA-Seq with rRNA and globin depletion. A) The number of detected genes per number of reads uniquely mapping to exons (FeatureCounts) was plotted for the samples shown in Figure x. B) Bar plot illustrating the number of detected genes for the samples shown in Figure x at 1 M reads /sample, normalized to Counts Per Million (CPM) at a threshold of CPM > 1.
Excellent Isoform Detection Across a Wide Range of Input Amounts
Lexogen’s Spike-in RNA Variant controls (SIRVs) are a set of 69 synthetic RNA molecules that mimic transcript isoform complexity. CORALL Total RNA-Seq V2 with RiboCop rRNA depletion authentically reproduces the expected coverage of the highly complex SIRV6 locus with 16 transcript isoforms and 2 anti-sense transcripts (Fig. 5). Thus, CORALL is ideal for isoform discovery and alternative splicing applications, even at inputs as low as 1 ng of total RNA (prior to ribo-depletion).
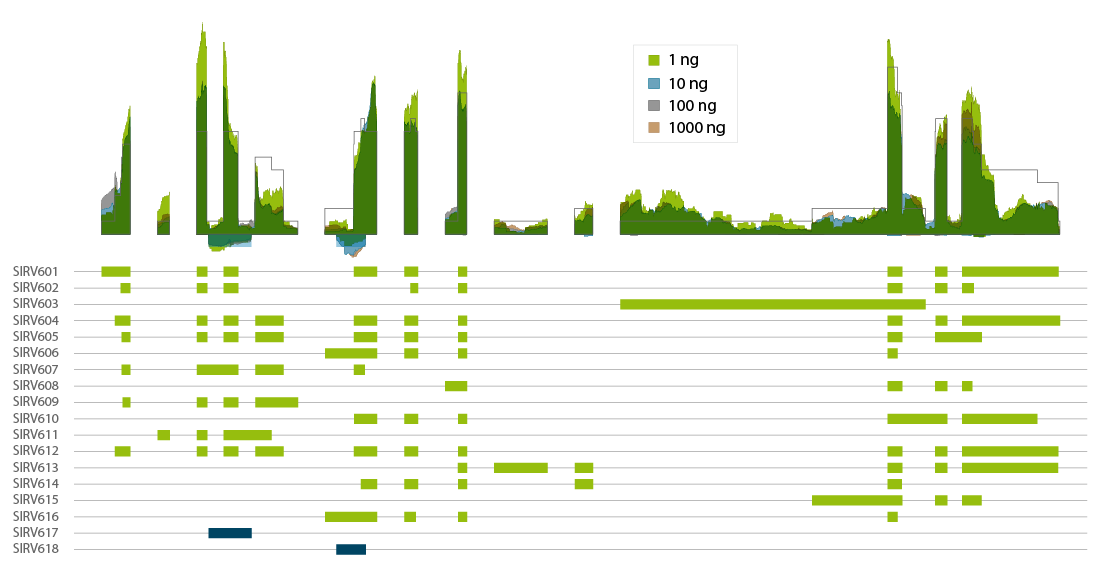
Figure 8 | Coverage transcript isoforms of SIRV6 across RNA input amounts. Lexogen’s SIRV-Set 3 was spiked into Universal Human Reference RNA at of 1 % of the mRNA fraction. Total RNA amounts of 1000 ng, 100 ng, 10 ng, and 1 ng were used for ribo-depletion with RiboCop and CORALL Total RNA-Seq V2 library preps for generation of libraries with long insert sizes. Libraries were sequenced on Illumina® NextSeq500 (2×150 bp). Reads were mapped to the SIRV reference genome and visualized in condensed coverage profiles on gene level (upper panel). The exon-intron structure of all 18 transcripts of the SIRV6 locus is shown in the lower panel (with antisense transcripts in blue). The coverage generated by CORALL Total RNA-Seq V2 is shown as overlay for four different input amounts.
Superior End-to-End Coverage
The innovative workflow of all CORALL versions delivers superior transcript start and end site coverage and exact representation transcription start sites.

Workflow
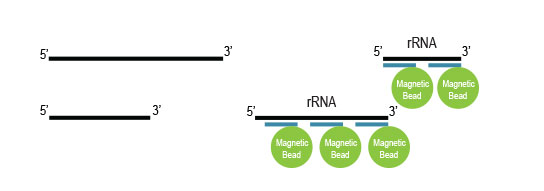
Samples are treated using a set of affinity probes for specific depletion of rRNA sequences and magnetic beads for depletion. RiboCop efficiently removes rRNA while maintaining unbiased transcript expression.

Following RiboCop, ribo depleted RNA can directly be used as template for CORALL reverse transcription. No prior RNA fragmentation is necessary, insert size is determined during reverse transcription.
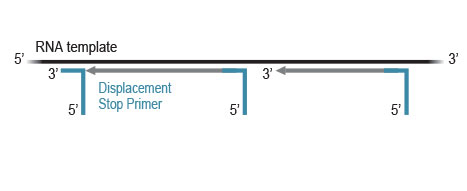
CORALL library generation is initiated by random hybridization of Displacement Stop Primers (DSP) with partial Illumina-compatible P7 sequences, to the RNA template. Reverse transcription extends each DSP to the next one, where transcription is effectively stopped. This stop prevents spurious second strand synthesis, maintaining
excellent strand specificity.
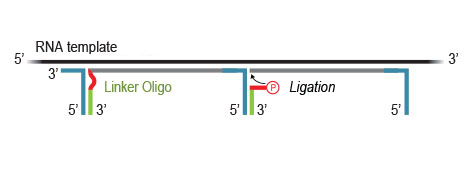
Highly efficient ligation of Linker Oligos to the 3’ ends of first-strand cDNA fragments then introduces partial Illumina compatible P5 sequences and Unique Molecular Identifiers (UMIs).

During PCR, second strand synthesis is performed, and the double-stranded cDNA is amplified. In doing so, unique dual i7 and i5 indices as well as complete adapter sequences required for cluster generation on Illumina instruments are added.
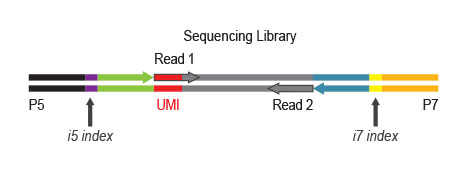
The UMI is positioned to be read out at the beginning of Read 1 during sequencing and can therefore be assessed even in Single-Read mode. All purification steps are based on magnetic beads, rendering the complete workflow highly suitable for automation.
FAQ
Frequently Asked Questions
Access our frequently asked question (FAQ) resources via the buttons below.
Please also check our General Guidelines and FAQ resources!
How do you like the new online FAQ resource? Please share your feedback with us!
Downloads
CORALL mRNA-Seq Library Prep Kit
Safety Data Sheet
If you need more information about our products, please contact us through support@lexogen.com or directly under +43 1 345 1212-41.
Ordering Information
| Cat. No. | Product Name |
| Stand-alone Kits without rRNA Depletion, including Lexogen 12 nt UDI Sets | |
| 171.24 | CORALL RNA-Seq V2 Library Prep Kit with UDI 12 nt Set A1, (UDI12A_0001-0024), 24 preps |
| 171.96 | CORALL RNA-Seq V2 Library Prep Kit with UDI 12 nt Set A1, (UDI12A_0001-0096), 96 preps |
| 172.96 | CORALL RNA-Seq V2 Library Prep Kit with UDI 12 nt Set A2, (UDI12A_0097-0192), 96 preps |
| 173.96 | CORALL RNA-Seq V2 Library Prep Kit with UDI 12 nt Set A3, (UDI12A_0193-0288), 96 preps |
| 174.96 | CORALL RNA-Seq V2 Library Prep Kit with UDI 12 nt Set A4, (UDI12A_0289-0384), 96 preps |
| 175.24 | CORALL RNA-Seq V2 Library Prep Kit with UDI 12 nt Set B1, (UDI12B_0001-0024), 24 preps |
| 175.96 | CORALL RNA-Seq V2 Library Prep Kit with UDI 12 nt Set B1, (UDI12B_0001-0096), 96 preps |
| 176.384 | CORALL RNA-Seq V2 Library Prep Kit with UDI 12 nt Sets A1-A4 (UDI12A_0001-0384), 384 preps |
| With rRNA Depletion, including Lexogen 12 nt UDI Sets | |
| 183.24 | RiboCop (HMR) and CORALL Total RNA-Seq V2 Library Prep Kit with UDI 12 nt Set A1, (UDI12A_0001-0024), 24 preps |
| 183.96 | RiboCop (HMR) and CORALL Total RNA-Seq V2 Library Prep Kit with UDI 12 nt Set A1, (UDI12A_0001-0096), 96 preps |
| 184.24 | RiboCop (HMR) and CORALL Total RNA-Seq V2 Library Prep Kit with UDI 12 nt Set B1, (UDI12B_0001-0024), 24 preps |
| 184.96 | RiboCop (HMR) and CORALL Total RNA-Seq V2 Library Prep Kit with UDI 12 nt Set B1, (UDI12B_0001-0096), 96 preps |
First time user of CORALL V2?
First Time User? We’re excited to offer you an exclusive introductory offer.
Buy from our Webstore
Not sure which CORALL RNA-Seq product is right for you?
Try the CORALL Configurator and find the ideal kit for your application
in less than 2 minutes!
Need a web quote?
You can generate a web quote by Register or Login to your account. In the account settings please fill in your billing and shipping address. Add products to your cart, view cart and click the “Generate Quote” button. A quote in PDF format will be generated and ready to download. You can use this PDF document to place an order by sending it directly to sales@lexogen.com.
Web quoting is not available for countries served by our distributors. Please contact your local distributor for a quote.



