Do you have any questions?
Is LUTHOR HD the right solution for your RNA research? Let’s find out!
LUTHOR HD
LUTHOR High-Definition Single-Cell 3′ mRNA-Seq Library Prep Kit
LUTHOR HD redefines the approach to single-cell RNA sequencing by offering an RNA-amplification-based kit able to detect virtually every single mRNA molecule present in the cell.
Like QuantSeq FWD, LUTHOR HD is a 3′ end mRNA-Seq kit that uses oligo(dT) primers to target polyadenylated RNAs and generate reads mapping to the 3′ end of transcripts. The two kits, however, use different protocols and technologies and are developed for different sample input amounts. While LUTHOR HD is designed for inputs ranging from 1 ng to 10 pg (equivalent to 100 cells to 1 cell) and even lower*, QuantSeq FWD is ideal for inputs ranging from 1 ng to 500 ng of total RNA.
LUTHOR HD is compatible with Lexogen’s patented 12 nt UDI Sets for maximal read rescue capabilities (no more misassigned or lost reads!).
* We have pushed the boundaries of sensitivity even further. LUTHOR HD enables to work with inputs as low as 1 pg, e.g. with cytoplasmic extracts. Discover more in our poster.
Did you know that CORALL and QuantSeq libraries built with UDI can be combined into a single flow cell, together with LUTHOR HD UDI libraries? For more information, please contact: support@lexogen.com or download our flyer here.
Performance
LUTHOR HD shows outstanding performance with ultra-low input samples – 10 pg – 1 ng (equivalent to 1 – 100 cells).
Up to 95% of genes detected at only 1M read depth
LUTHOR HD brings unparalleled sensitivity, detecting the majority of expressed genes at only 1 M read depth, both with isolated single cells and with equivalent amounts of purified RNA (Fig. 1).
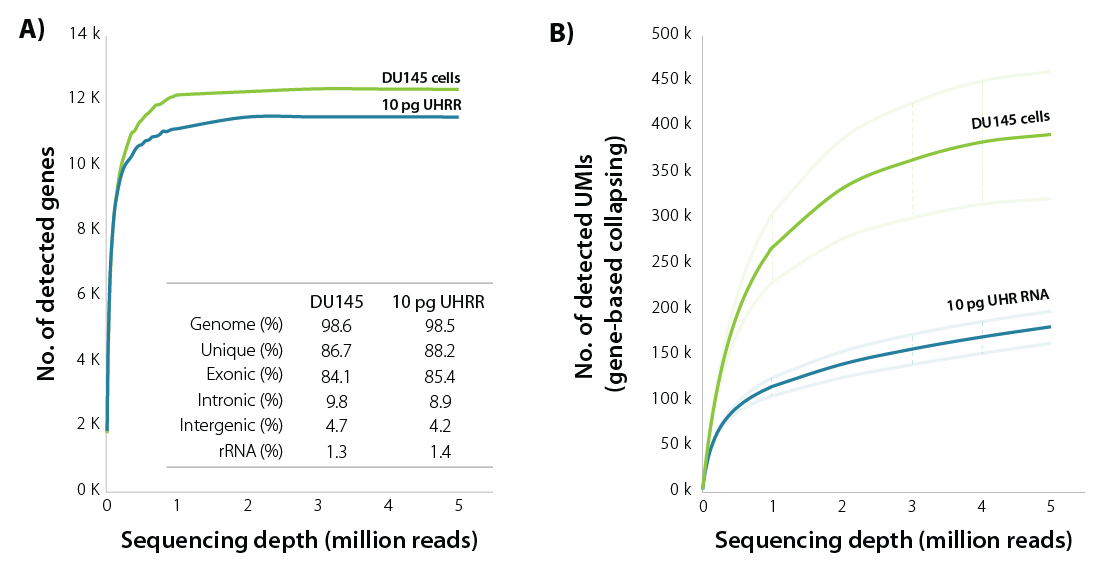
Figure 1 | Sensitivity of LUTHOR HD. A) Scatter plots of the average number of genes detected per DU145 human cell (contains 18.3 ± 1.5 pg of total RNA) and 10 pg Universal Human Reference RNA (UHRR), inferred across four replicates at stepwise-reduced read fractions (CPM > 1). Inset table shows sequencing alignment metrics across four DU145 and four 10 pg UHRR replicates at 1 M read depth. Variability between replicates is negligible (not shown here).
B) Scatter plots of the average number of UMIs (UMI = 12 bp) detected per DU145 human cell and 10 pg UHRR, inferred across four replicates at stepwise-reduced read fractions. UMI counts represent all unique alignments after gene-based collapsing. The number of transcripts directly correlates with cell size (large cells have more transcripts, small cells have fewer transcripts).
Outstanding sequencing statistics - minimal rRNA and gDNA reads
Whether you work with single isolated cells or with larger pools of 10 or 100 cells, your read statistics will consistently provide you with very high exonic coverage. rRNA and gDNA will not interfere with your results.
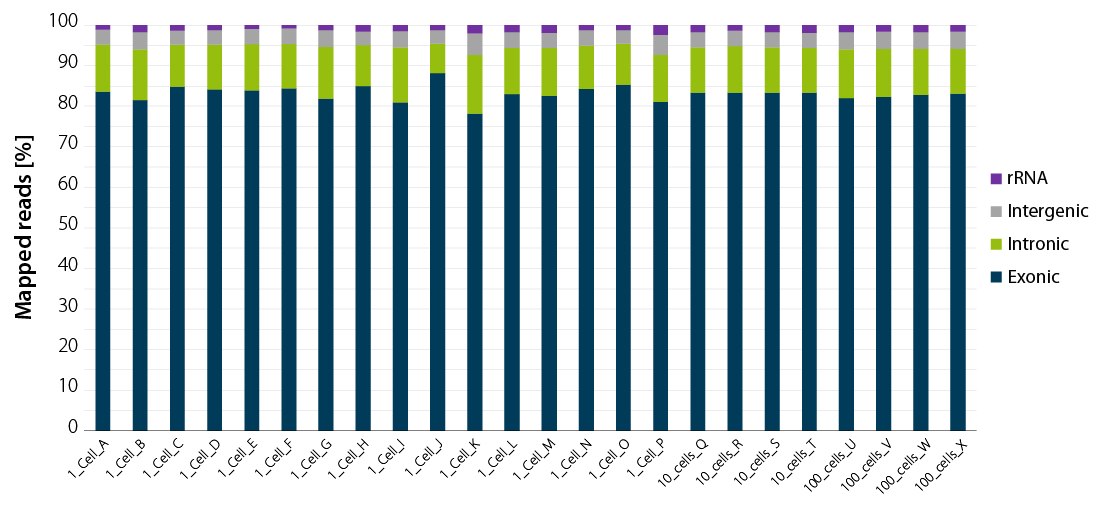
Figure 2 | LUTHOR HD NGS read statistics. Horizontal stacked bar graph shows sequencing alignment metrics across 16 single DU145 cells (named A to P), 4 groups of 10 cells (named Q to T), and 4 groups of 100 cells (named U to X), freshly isolated by flow-cytometry-assisted sorting and immediately lysed in LUTHOR HD Cell Lysis Buffer (CLB). Read depth per library ranges from 0.44 to 1 M reads (1 cell), 0.78 to 1 M reads (10 cells) and 0.80 to 1 M reads (100 cells). Total mapped reads percentages are split into exonic, intronic, intergenic, and ribosomal RNA reads.
High reproducibility, even at ultra-low input
At Lexogen, we have built a kit that consistently delivers very high reproducibility. Even if the experiment is repeated by different operators.
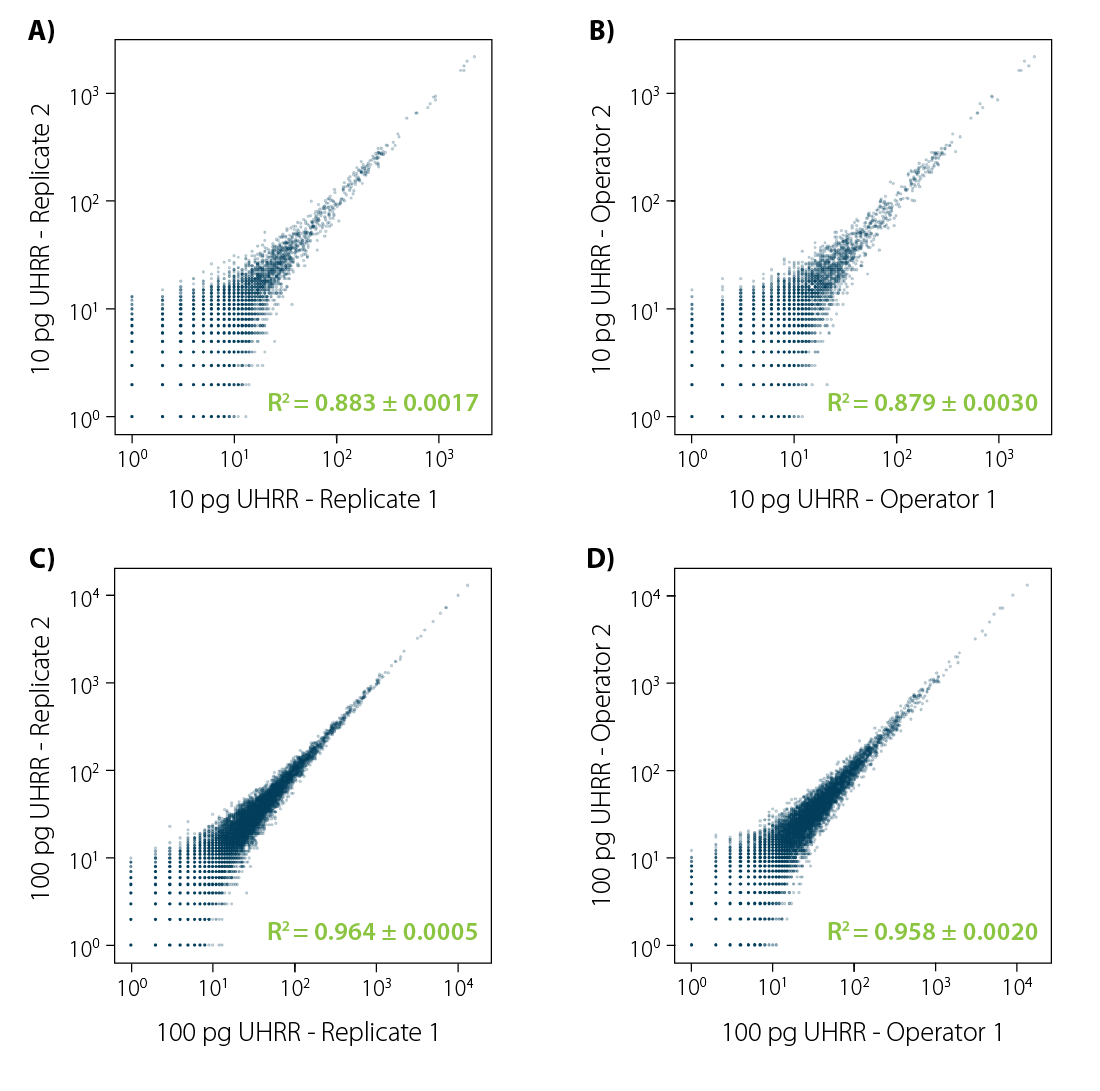
Figure 3 | LUTHOR HD reproducibility. Correlation plots (orthogonal regression of gene-based collapsed and mapped reads) obtained from UHRR reference samples. Four different comparisons were conducted:
A) same operator, 4 technical replicates (6 comparisons), 10 pg input;
B) two different operators, 4 technical replicates per operator (16 comparisons);
C) same as A, with 100 pg input;
D) same as C, with 100 pg input. Each image illustrates an exemplary comparison of two samples. Variance (±) indicated for the R² value is the result of all combinations in each comparison experiment.
Workflow
Single, e.g. FACS-sorted animal cells or ultra-low input RNA (10 pg -1 ng)
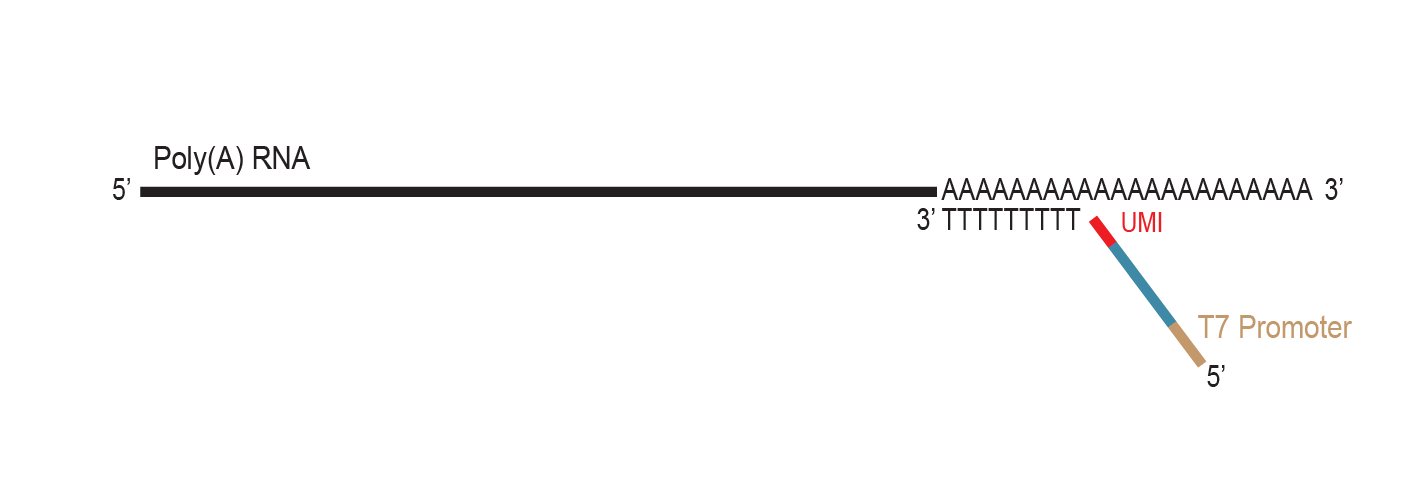
The primer contains Unique Molecular Identifiers (UMI, in red), a partial Illumina compatible P7 linker (blue), and a T7 promoter sequence (light brown).
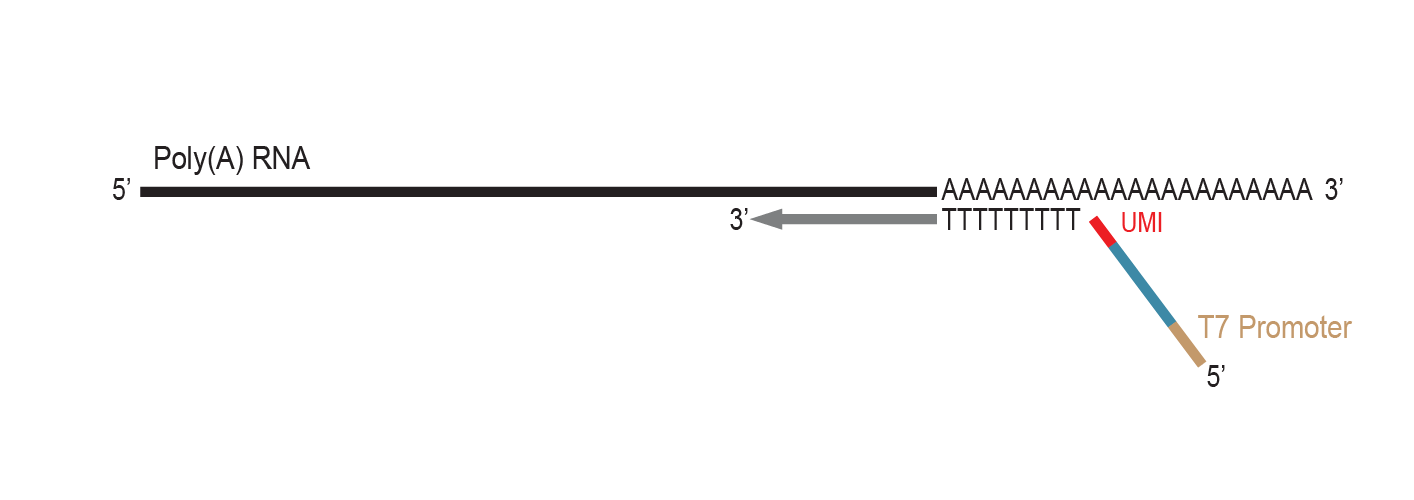
Reverse transcriptase elongates the primer and generates cDNA to stabilize the RNA template.
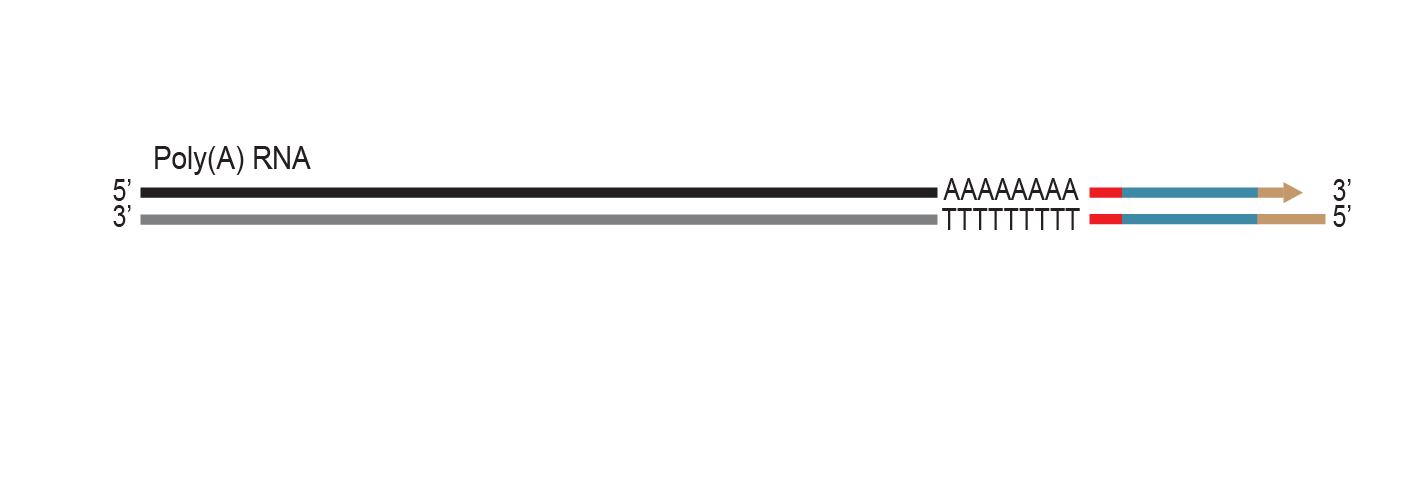
The proprietary end repair step removes the single-stranded 3’ poly(A) overhang and generates a double stranded T7 promoter
sequence for RNA amplification.
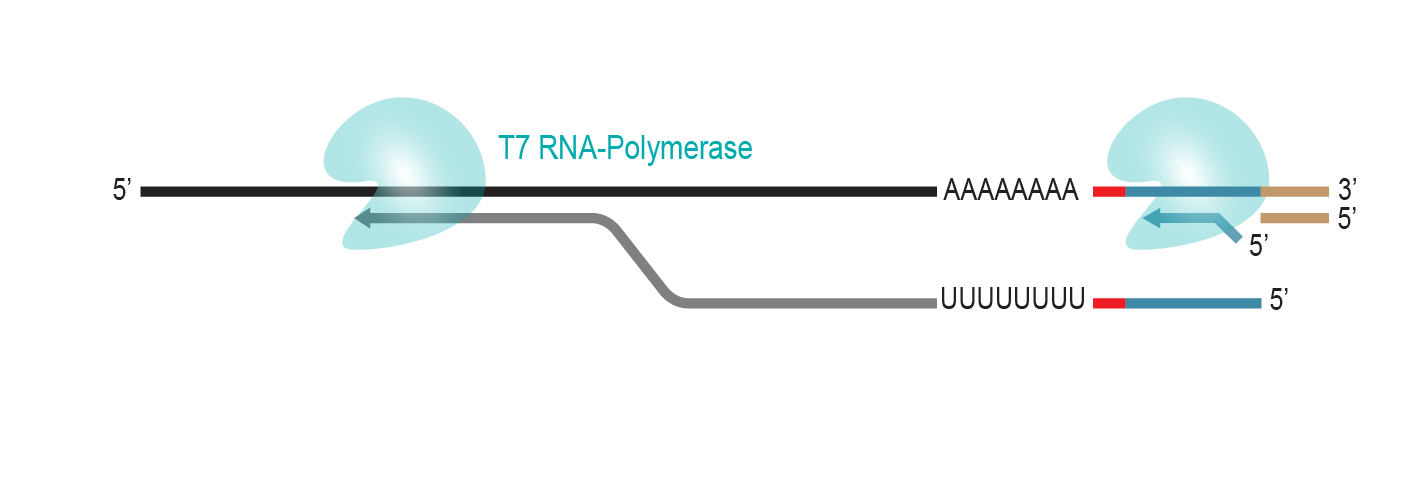
During a 2-hour (optional: overnight) in vitro transcription step the original mRNA template is copied repeatedly by linear amplification.
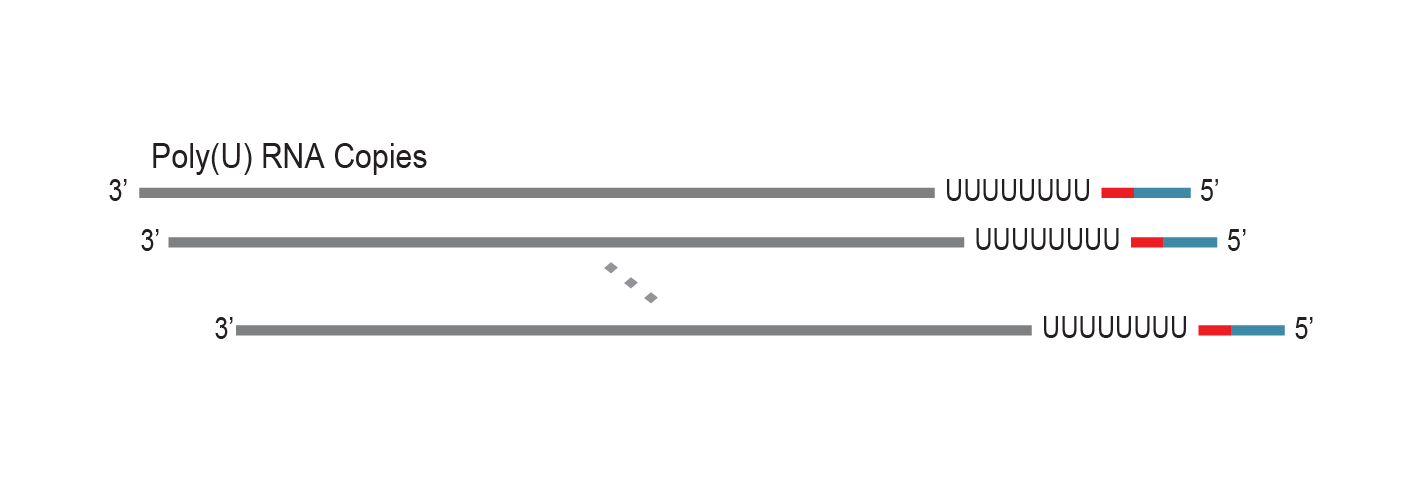
Antisense-RNA copies are generated which contain the partial Illumina compatible P7 linker at the 5’ end.
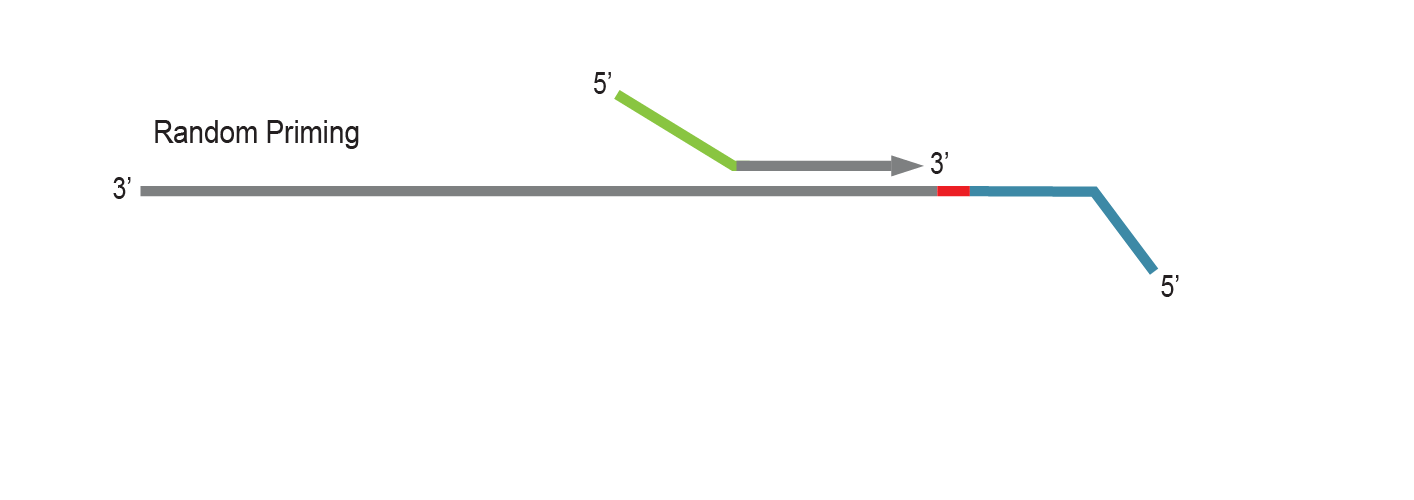
LUTHOR library generation is initiated by random priming using Lexogen’s proprietary Displacement Stop technology. Random primers contain partial Illumina-compatible P5 sequences (green).
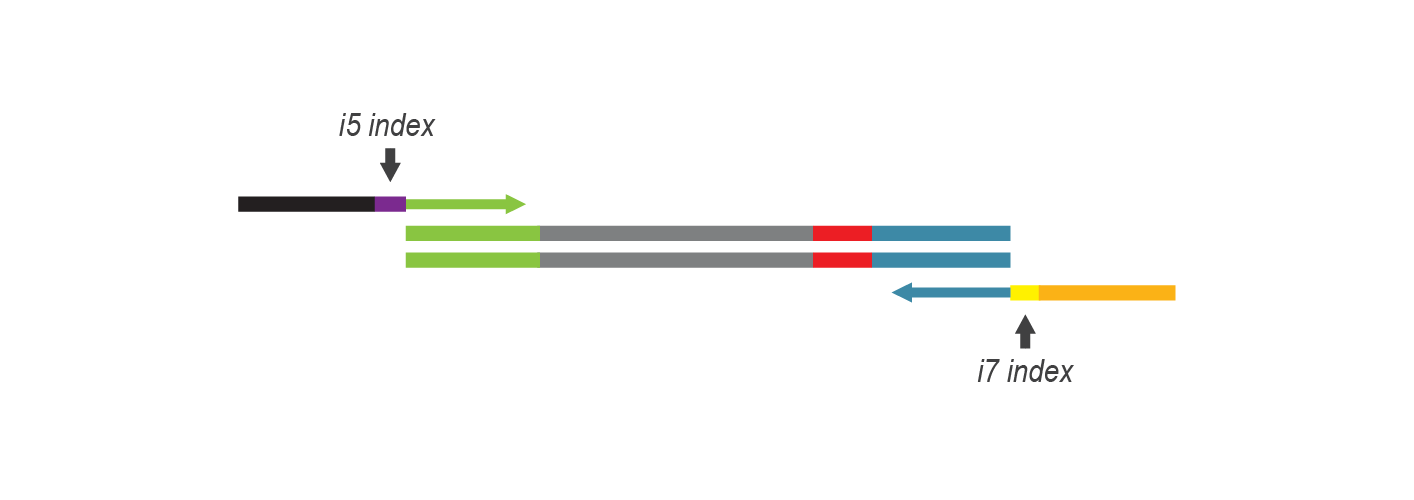
During PCR, second strand synthesis is performed, and the double-stranded cDNA is amplified. In doing so, i7 and i5 indices as well as complete adapter sequences required for cluster generation on Illumina instruments are added.
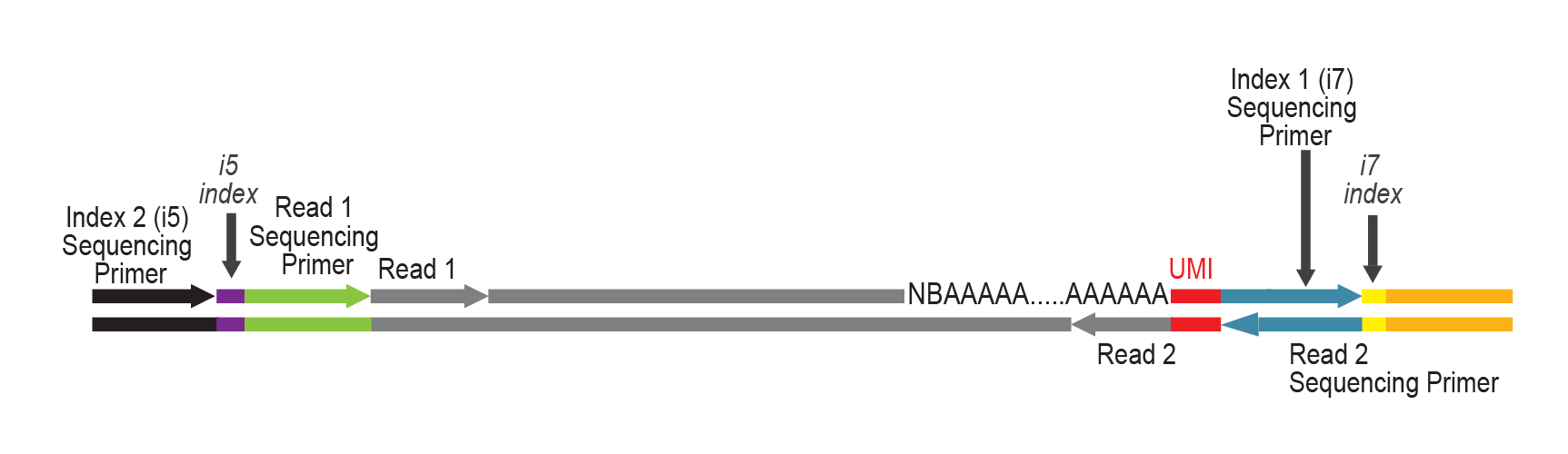
LUTHOR contains the Read 1 linker sequence in the random primer, hence NGS reads are generated towards the poly(A) tail and directly correspond to the mRNA sequence. LUTHOR libraries should be sequenced in asymmetric PE mode: Read 1 for the insert, Read 2 for the UMI only (12nt).
Data Analysis on Kangooroo
Each LUTHOR HD kit includes a voucher code for complimentary data analysis on Kangooroo - Lexogen’s web-based interactive RNA-Seq analysis platform. Analyse your data quickly and conveniently - no coding skills are required.
FAQ
Frequently Asked Questions
Access our frequently asked question (FAQ) resources via the buttons below.
Please also check our General Guidelines and FAQ resources!
How do you like the new online FAQ resource? Please share your feedback with us!
Downloads
LUTHOR 3′ mRNA-Seq Kit
Safety Data Sheet
If you need more information about our products, please contact us through support@lexogen.com or directly under +43 1 345 1212-41.
Ordering Information
| Cat. No. | Product Name |
| LUTHOR HD without UDI | |
| 204.24 | LUTHOR High-Definition Single-Cell 3′ mRNA-Seq Kit, 24 preps |
| 204.96 | LUTHOR High-Definition Single-Cell 3′ mRNA-Seq Kit, 96 preps |
| LUTHOR HD with UDI | |
| 221.24 | LUTHOR High-Definition Single-Cell 3′ mRNA-Seq Kit with UDI 12 nt Set B1, (UDI12B_001-0024), 24 preps |
| 221.96 | LUTHOR High-Definition Single-Cell 3′ mRNA-Seq Kitwith UDI 12 nt Set B1, (UDI12B_0001-0096), 96 preps |
First time user of LUTHOR?
First Time User? We’re excited to offer you an exclusive introductory offer.
Buy from our Webstore
Need a web quote?
You can generate a web quote by Register or Login to your account. In the account settings please fill in your billing and shipping address. Add products to your cart, view cart and click the “Generate Quote” button. A quote in PDF format will be generated and ready to download. You can use this PDF document to place an order by sending it directly to sales@lexogen.com.
Web quoting is not available for countries served by our distributors. Please contact your local distributor for a quote.