The roots of classical single-cell RNA sequencing (scRNA-Seq) trace back to the desire and need to capture the nuances of individual cells within a heterogeneous cell population. Early efforts laid the foundation for breakthroughs in gene expression profiling, paving the way for technologies like scRNA-Seq to emerge as powerful tools in the biological researcher’s arsenal.
Single-cell transcriptomics began with the development of single-cell qPCR. Then, based on the advances made in single-cell qPCR, whole-transcriptome analysis was performed using microarrays, and subsequently (bulk) RNA sequencing (RNA-Seq) was adapted for the analysis of single cells, and scRNA-Seq was born (Kolodziejczyk et al., 2015).
scRNA-Seq is a revolutionary technique that allows researchers to delve into the fascinating world of cellular complexity and explore gene expression dynamics on a cell-by-cell basis. The first transcriptomes generated by scRNA-Seq were published by Tang et al. in 2009 (Tang et al., 2009), just 2 years after the first applications of RNA-Seq to bulk cell populations. Since its initial discovery, scRNA-Seq has provided insights into transcriptional profiles of cells in various biological research fields, leading to exciting new discoveries in better understanding the composition and interaction of cells in humans, model animals and plants.
In this blog post, we will provide an overview of the basic principles, benefits, and most common applications of classical single-cell RNA-Seq.
What does scRNA-Seq tell us that bulk RNA-Seq cannot?
Complex biological systems result from the coordinated functions of individual cells, each of which contributes its own unique role to the organism as a whole. Because of this complexity, gene expression research in organisms, tissues or cell populations is often limited by traditional bulk RNA-Seq methods, and single-cell RNA-Seq approach is needed.
To understand the difference between the capabilities of scRNA-Seq and bulk RNA-Seq, it is helpful to use a smoothie analogy. Imagine a smoothie made from a variety of fruits, vegetables, and herbs as a metaphor for what bulk RNA-Seq provides, a blended average of gene expression from a diverse population of cells. Now imagine using scRNA-Seq as a tool to break down this complex smoothie into its individual components. Instead of sipping the entire blend, you can now know the exact amount of strawberries, identify the nuances of each ingredient, and even spot the occasional outlier like a lone blueberry or taste a dash of cinnamon.
By isolating and analyzing individual cells, scRNA-Seq unlocks the richness of cellular heterogeneity within a tissue or organ, allowing a closer look at transitional states and subtle variations in gene expression. It is like having the ability to zoom in on the unique genetic signature of each cell, revealing rare cell types that might be missed in a bulk analysis. This approach is particularly valuable when studying tissues with diverse cell populations, where bulk RNA-Seq can provide a “blended”, “mixed” set of information that masks the distinct characteristics of specific cell types. While bulk RNA-Seq gives us a broad overview, scRNA-Seq allows researchers to appreciate the individual flavours within the cellular landscape (Figure 1).
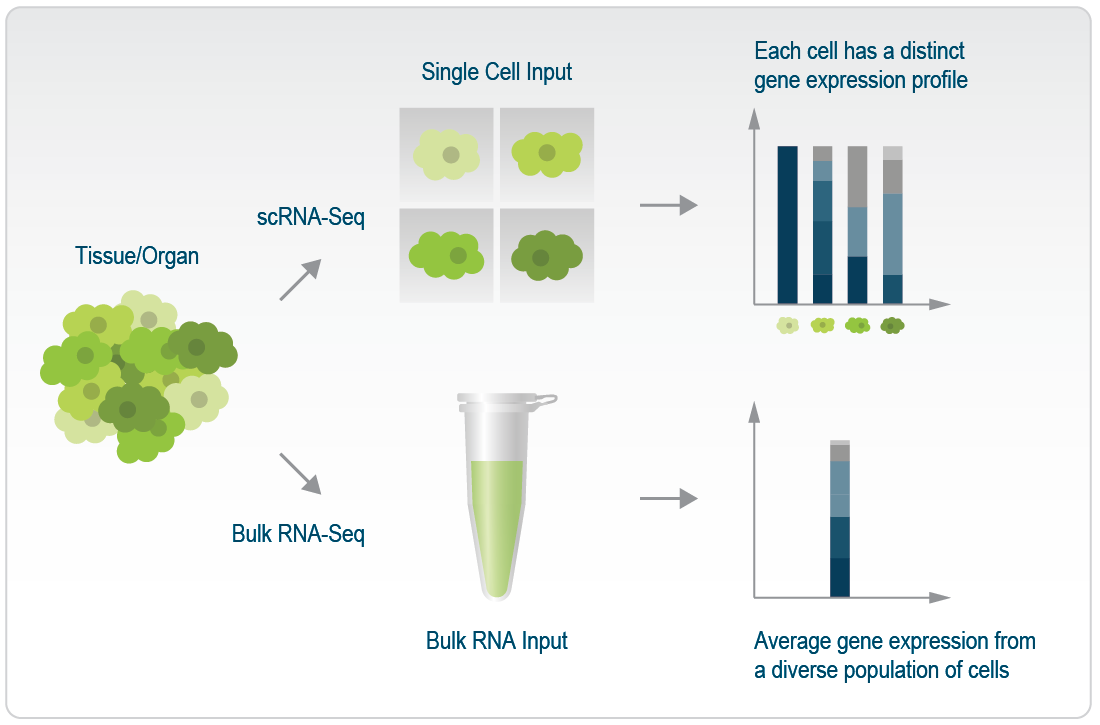
Figure 1 | Difference in the gene expression profiles between scRNA-Seq and bulk RNA-Seq
Why and when should you consider performing scRNA-Seq?
scRNA-Seq enables transcriptional profiling of thousands of individual cells and allows us to describe RNA molecules in individual cells at high resolution. This high-throughput analysis will help answer important questions, such as:
- Which genes are expressed and at what levels at the single cell level?
- How do transcriptional profiles differ across thousands of cells in a heterogeneous sample?
- What cell types are present in the sample?
- How do individual cells contribute to the function of complex biological systems?
- How do the transcriptional profiles of cells change during development, or in a course of a treatment?
From the earliest days of scRNA-Seq, it was clear that the method could reveal transcriptional similarities and differences within a population of cells, enabling the discovery of previously unrecognized levels of heterogeneity in, for example, embryonic and immune cells – and to this day, the analysis of cell heterogeneity is a core reason for embarking on scRNA-seq studies (Haque et al., 2017). In line with this, the assessments of transcriptional differences between individual cells have been used to identify rare cell populations that would otherwise go undetected in analyses of pooled cells. Especially in cancer research, scRNA-Seq has become irreplaceable in identifying rare tumor cells, e.g., malignant tumor cells within a tumor (Tirosh et al., 2016)or hyper-responsive immune cells (Shalek et al., 2014) within a seemingly homogeneous group. scRNA-seq is also ideal to look at single cells where each one is essentially unique, such as individual T lymphocytes expressing highly diverse T-cell receptors, or neurons, or cells within an early-stage embryo (Haque et al., 2017). We will talk more about specific applications of scRNA-Seq under “Common Applications of Single-Cell Sequencing“.
How Does Classical Single-Cell Sequencing Work?
It is important to note that while scRNA-Seq can provide answers to many research questions, the details of the answers provided will vary depending on the protocol used. Different protocols for scRNA-Seq will dictate the level of detail that can be resolved from the mRNA data, such as how many genes/transcripts of each gene can be detected, whether a particular gene of interest is expressed, whether differential splicing has occurred, etc (Ziegenhain et al., 2017; Chen et al., 2019). In our next blog posts, we will look in more detail at comparing different protocols and what they can provide in terms of depth of information, and here we will outline the main steps required for scRNA-Seq.
Single-cell RNA sequencing follows a well-established process (which we here divide into five main steps), regardless of the sequencing platform or scRNA-Seq technology you use.
Step 1: Generate a single cell suspension.
To isolate single cells from a population of cells, one must first generate a single cell suspension. Typically, we start with the primary tissue or organ of interest and apply some of the methods for tissue dissociation. Common techniques rely on enzymatic digestion and/or the application of some type of mechanical force to the tissue. It is important to note that extracting single cells from tissues is not always an easy task, so we would recommend that before embarking on a scRNA-Seq journey, researchers should optimize the recovery of single cells from the target tissue to avoid excessive alteration of the transcriptome during the procedure.
Step 2: Isolate the cells.
Once cells are in a single-cell suspension, there are several ways how to isolate them for scRNA-Seq, and the most common being fluorescence-activated cell sorting (FACS), magnetic-activated cell sorting, microfluidic system and laser microdissection.
FACS sorting is a very simple approach that uses a flow cytometer (FACS) machine to sort live cells into the wells of a 384-well microtiter plate. Live/dead staining is included to discard dead cells and sort only live cells. FACS is particularly useful when only a specific cell type from the tissue/organ is of interest – by using marker-specific fluorescently labeled antibodies, only cells expressing the desired marker are sorted onto the plate.
Technologies using microfluidics are also very common, and here sorting is done using a droplet-based microfluidics approach, where a single cell suspension is loaded onto the chip, which contains beads for barcoding and other necessary reagents. By applying pressure to the cell suspension, the cells are forced one by one through tiny capillaries in the chip and as a result separates them into droplets. In the next step, the droplet containing the single cell is combined with a droplet containing barcodes and reagents ready for step 3.
Step 3: Extraction, processing and amplification of the genetic material of each isolated cell.
One of the first challenges in developing scRNA-Seq as a method was finding a way to measure the tiny amount of RNA contained in a single cell. In 2009, researchers succeeded in amplifying the RNA from a single cell to the point where it could be sequenced for the first time (Tang et al., 2009).
Today, amplification is done by PCR and/or in vitro transcription (IVT), using primers that anneal to the mRNA.
Regardless of the technology used, the cells must first be lysed (so that the RNA content remains), followed by a step of mRNA capture and reverse transcription. To specifically capture polyadenylated mRNA molecules and avoid capturing ribosomal RNAs, poly(T) primers are commonly used. Poly(T)-primed mRNA is converted into complementary DNA (cDNA) by a reverse transcriptase, and depending on the scRNA-Seq protocol, other nucleotide sequences are added to the reverse transcription primers, such as adaptor sequences for detection on NGS platforms, unique molecular identifiers (UMIs) to uniquely mark a single mRNA molecule, and sequences to preserve information about the cellular origin (barcodes) (Haque et al., 2017). After second-strand synthesis, the minute amounts of cDNA are then amplified either by PCR or, in some protocols, by in vitro transcription (IVT), followed by another round of reverse transcription – some protocols opt for barcoding at this stage to preserve information about the cellular origin. This barcode takes the form of a short nucleotide sequence that is incorporated into the amplification primers and allows individual cells to be distinguished during data analysis.
Step 4: NGS library preparation and sequencing.
After amplification, amplified and tagged cDNA from every cell is pooled and sequenced by NGS. Library preparation techniques, sequencing platforms and genomic-alignment tools are similar to those used for bulk RNA-Seq (Haque et al., 2017; Jovic et al., 2022).
Step 5: Data analysis.
Bioinformatic methods are used to assess quality and variability, as well as to interpret and present the data. The analysis of single-cell datasets presents a number of specific challenges, such as dealing with sparsity (i.e., scRNA-Seq datasets have large fractions of observed zeros, where a given gene in a given cell has no unique molecular identifiers or reads mapping to it), defining statistical frameworks for discovering complex differential patterns in gene expression, correctly mapping single cells to a reference atlas, and generalizing trajectory inference (i.e., determining the pattern of a dynamic gene expression), and finding patterns in spatially resolved measurements.
If you are interested in diving deeper into the current status, problematics and challenges in analyzing single-cell RNA-Seq datasets, we would recommend looking into the review “Eleven grand challenges in single-cell data science” by Lähnemann, D., et al. published in Genome Biology.
Common Applications of Single-Cell Transcriptomics
Single-cell RNA-Seq has become indispensable in fields such as developmental biology, immunology, neuroscience, and cancer research – as it can be used for answering most biological questions that require a detailed understanding of cell populations. Researchers are harnessing its power to explore cellular diversity in tissues, track immune responses at the single-cell level, decipher neural pathways, and unravel the complexities of cancer development. Let’s take a look at some of the most exciting applications and discoveries made possible by single-cell transcriptomics.
Using single-cell transcriptomics to map cells in entire organisms
Single-cell transcriptomics was first applied to several model organisms, and is still crucial for understanding the interplay between cells in mouse, zebrafish, worms, etc. Two high-throughput collections of information on mouse tissues profiled by scRNA-Seq have been published, providing data on many tissues using standardized protocols, providing a biological resource of great value (The Tabula Muris Consortium, 2018; Han et al., 2018).
The average adult human has 37 trillion cells, but we still do not know how many different cell types there are, their properties in health and disease, nor how they work together. From it’s first use, technological developments and protocol improvements have driven a consistent exponential increase in the numbers of cells studied in scRNA-Seq analyses, from tens of cells up to hundreds of thousands single cells per study. This so-called scaling up has enabled scRNA-Seq technology to be used to map the entire human body, in an impressive undertaking by the researchers gathered around the Human Cell Atlas.
The Human Cell Atlas is a global consortium led by Sarah Teichmann and Aviv Regev, in which scientist are using high-throughput technologies at the single-cell resolution to map all the cells in human tissues and organs, creating a kind of “Google maps” of the human body. Creating a reference atlas of both human tissues and model organisms in health is a prerequisite for observing changes in ageing, disease and in response to treatments. This admirable undertaking aims to provide an openly available, globally representative resource, that will enable researchers worldwide to study and understand health and disease. The more detailed goals of the initiative can be found in the About section of the Consortium’s webpage. The webpage also contains a variety of resources, including publications, educational videos and training materials.
Single-cell RNA-Seq in development
Cell mapping of human tissues has been especially significant in fuelling fascinating studies of human development, including embryogenesis, organogenesis, but recently also reproductive biology and prenatal genetic diagnosis. Single-cell atlases of development are now available for organs and systems including brain, heart, liver, thymus, gut, and kidney, and recently, the first single-cell atlas of the maternal-fetal interface in early pregnancy (Vento-Tormo et al., 2018). This remarkable study revealed new cell states, and a more detailed cellular and molecular understanding of the complex architecture of this “shared” tissue and its role in maternal immune tolerance. In addition, the study revealed mechanisms behind healthy pregnancy and aided in understanding of how these could change resulting in conditions such as pre-eclampsia, a dangerous complication of pregnancy. Another capital study focused on the human thymus, the organ responsible for the maturation of many types of T cells, which are immune cells that protect us from infection. What was not well known is how these cells develop with a full immune complement that contains the necessary variation to protect us from a variety of pathogens. By performing scRNA-Seq on thymic cells, Park et al. examined the changes that occur in the thymus over the course of a human life. They could learn that development occurs in a coordinated manner among immune cells and with their developmental microenvironment, allowing for the creation of models of how T cells with different specific immune functions develop in humans (Park et al., 2020).
Single-cell RNA-Seq in cancer research
scRNA-Seq has become the state-of-the-art approach to study the heterogeneity and complexity of transcripts between individual cells, which is particularly helpful in addressing the heterogeneity of cancer. scRNA-Seq is being used for tumor prevention, diagnosis, and even treatment. It has found tremendous application in the study of tumorigenesis, allowing us to gain insight into the genetic and molecular characteristics of different stages of tumor development, from precancerous lesions to invasive tumors and metastasis. In patients with metastatic disease, personalized approaches such as targeted immunotherapy have proven to be the most effective. Performing scRNA-Seq on metastatic tumor cells can help analyze the immune heterogeneity of tumors and help design personalized, targeted immunotherapy (Lei et al., 2021; Zhang et al., 2021; Ergin et al., 2022).
Mapping and understanding cancer heterogeneity
Bulk RNA-Seq technologies detect all RNA molecules in a sample, thus not being able to provide information about gene expression patterns of individual cells. Tumor heterogeneity plays an important role in cancer progression and understanding gene expression patterns in different “subpopulations” within a tumor is a prerequisite to understanding cancer progression (Ergin et al., 2022). This is where scRNA-Seq comes into play and solves the issue by providing single-cell resolution of the transcriptome. scRNA-Seq was used in melanomas to analyze over 4,000 cells isolated from tumors of several patients, including cancer cells, immune cells, endothelial cells, etc. Data pointed to a vast heterogeneity of cells within the same cancer, being associated with cell cycle, spatial background, and resistance to drugs (Tirosh et al., 2016).
While scRNA-Seq has been useful in understanding many cancer types’ heterogeneity, this method has some limitations that prevent its application to all cancer types. The primary obstacle is that scRNA-Seq methods require single cells to be isolated from tissues while remaining intact and viable, which is often challenging, as in the case of extracting undamaged neurons from the brain. In this case, spatial transcriptomics provides a solution (Ergin et al., 2022). It eliminates the need to isolate cells from the tissue, as transcriptomics is performed in situ. Additionally, having knowledge of spatial information has proven to be very helpful – studies have shown a high heterogeneity in differential gene expression among different foci within one tumor.
Another method, single-nucleus RNA-Seq (snRNA-Seq), is also being used to study cancer heterogeneity, especially when dealing with samples that are a limitation to using scRNA-Seq (e.g., frozen tissues). snRNA-Seq profiles gene expression using isolated nuclei instead of entire cells and, compared to scRNA-Seq, is better suited for profiling gene expression in cells that are difficult to isolate, such as adipocytes and neurons, as well as cells in preserved tissues. Nuclei can be extracted from fresh, lightly fixed, or frozen tissues, which helps obtain less disturbed/altered transcriptomes during isolation. However, as nuclei have lower amounts of mRNA compared to cells and are more challenging to enrich or deplete for specific cell types of interest, these experiments require different optimizations and customizations (Wu et al., 2019; Slyper et al., 2020).
Studying cancer microenvironment and immunotherapy
The immune system influences cancer development, tumorigenesis, progression, and metastasis. Tumor-infiltrating lymphocytes (TILs), as their name suggests, infiltrate tumors and have a rich interplay with malignant and stromal cells. The cancer microenvironment, an ecosystem that surrounds tumors in the body, includes immune cells such as TILs, blood vessels, cells that make up connective tissue, and so on. A tumor and its microenvironment are in constant interaction, and the tumor microenvironment can either positively or negatively influence tumor development, progression, and metastasis. Therefore, as important as it is to study cancer cells, it is equally important to study all the cells in the tumor microenvironment (Tirosh et al., 2016; Zhang et al., 2021; Ergin et al., 2022).
scRNA-Seq has found its greatest application in immunology, making it an ideal method to study cancer microenvironment, which is highly enriched in different types of immune cells. The data obtained by scRNA-Seq revealed a huge heterogeneity of immune cell subpopulations in cancer tissues, and by deepening the understanding of the interplay between immune cells and cancer cells, provided a whole new insight into possible anti-cancer immunotherapies, opening up new avenues for cancer therapy (Zhang et al., 2021)(4).
When T cells are exposed to inflammatory signals, as in the case of cancer, they can deteriorate or become “exhausted.” Exhausted T cells lose robust effector functions, express multiple inhibitory receptors, and are defined by an altered transcriptional program, making them inefficient in cancer control. scRNA-Seq has, for example, helped identify transcription factor NFATC1 as a potential molecular signature of T cell exhaustion in metastatic melanoma (Tirosh et al., 2016).
Immunotherapies have revolutionized anti-cancer treatment, but there is still a huge variety in how individual patients respond to immunotherapy. Many unknowns are still in the way of immunotherapies being fully successful. scRNA-Seq is a vital tool in these endeavors to understand cancer better. Subpopulations of cells with persistence to immunotherapy have been identified in melanoma patients thanks to scRNA-Seq. For patients who are not responding to immunotherapy, scRNA-Seq may be able to analyze specific tumor tissues and elucidate the underlying mechanisms of resistance. scRNA-Seq has also been used to explore the combinatorial effect of immunotherapy, chemical, or radiation therapy to improve clinical effects among patients, while limiting the side-effects of therapies (Zhang et al., 2021).
In 2020, the Human Tumor Atlas Network was proposed by the joint forces of several world-leading cancer research centers. 3-dimensional atlases of cancer transformation, including precancerous lesions and primary and metastatic tumors, have been assembled based on scRNA-Seq data, combined with anatomy, histology, spatial information, proteome, epigenome data, and microbiomes. Through comparative analyses of these atlases, one can get a much more detailed picture of cancer heterogeneity and treatment resistance in different patients, during different periods, compared to when using only genomic data. This approach showed very nicely how the right way in cancer research, prevention, and therapy is to join forces and apply multi-disciplinary approaches whenever possible.
Single-cell RNA-Seq in immunology
The immune system is a complex, dynamic, and plastic system composed of many different types of immune cells that constantly sense and interact with their local microenvironment to protect us from infection or from our own pathologies (e.g., cancer cells) with the goal of maintaining homeostasis. Much effort and creativity has been devoted to the characterization of immune cells and their microenvironment, but traditional marker-based and bulk technologies have left many key questions unanswered. Since the advent of single-cell genomics, our knowledge of the cellular and molecular makeup of the immune system has blossomed, reshaping immunological research as we know it (See et al., 2018; Cao et al., 2020).
Starting with the use of scRNA-Seq to map mouse spleen immune cells, researchers began to use the method to get new insights into classical immunological models and questions, such as hematopoiesis, to study the interplay between immune cells, cancer and stromal cells (see more in Single-cell RNA-Seq in cancer research), to dissect molecular drivers of immune responses in combination with CRISPR-CAS9 technology (Dixit et al., 2016; Jaitin et al., 2016; Shaheen et al., 2018), and to dive into neuroinflammation and explore neurodegradation in a study that identified an unique microglia type associated with the development of Alzheimer’s Disease (Keren-Shaul et al., 2017). After these initial, but groundbreaking studies, momentum built and soon there was an explosion of applications of scRNA-Seq (and discoveries) in immunology, with new publications appearing all the time, shedding more and more light on the heterogeneity of the immune system, rare immune cell types, and the nuances of acute and chronic inflammation, autoimmunity, and anti-tumor immune activity. For an excellent overview on past, present and future of single-cell immunology, we recommend looking into this comprehensive review by Ginhoux et al, published in Immunity.
Single-cell RNA-Seq in diabetes
scRNA-seq technology is being used to advance the understanding of pancreatic β-cell development and pathology in diabetes. The cure for type 1 diabetes (T1D) lies in restoring β cells, but to generate functional β cells, we need to understand pancreatic development, molecular events, and knowledge of cellular heterogeneity in health and disease. To date, scRNA-Seq has been used to study the developing pancreas, first in mouse models and then in human pluripotent models, mainly 3D models of embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). These studies have shed new light on pancreatic development, identifying novel cell markers, discovering ß-cell heterogeneity and transcriptomic dynamics during postnatal maturation and proliferation, and characterizing the mechanisms behind pancreatic β- and α-cells, generation, expansion, and maturation during pancreatic development (Stanescu et al., 2017).
The current state-of-the-art in the field is to use 3D organoids as a patient-specific cell model to study the transcriptome of T1D, and scRNA-Seq is an important method for looking into individual cells’ transcriptomes. However, many challenges stand in a way of these experiments due to the high hydrolytic enzymatic activity of pancreatic cells, which makes it difficult to obtain good material for scRNA-Seq. Employing single-nucleus RNA-Seq on frozen pancreas, instead of scRNA-Seq, is one of the solutions (Jovic et al., 2022).
Single-cell RNA-Seq in neurobiology
The central nervous system (CNS) has served as an excellent playground for single-cell technologies since their inception. The high cellular complexity and “individualistic” nature of neurons provided an ideal study object for the increasing power of various single-cell methods, including scRNA-Seq, which became an irreplaceable tool for studying the molecular and cellular repertoire of the CNS and its many disorders. However, scRNA-Seq requires the dissociation of tissue samples, which means that the interrelationships between cells are lost – so spatial transcriptomic methods are often used to overcome this challenge. Some of the areas of neurobiology where scRNA-Seq has provided the most insight are studies of selective neuronal vulnerability, neuroimmune dysfunction, expanding understanding of pathomechanisms of brain disorders and cell type-specific treatment response.
As usual, mouse models were used for the initial studies (Zeisel et al., 2015), such as the one in which transcriptomes of more than 3,000 individual cells from the mouse somatosensory cortex and hippocampal CA1 region were analyzed by scRNA-Seq, followed by a study in which the transcriptomic profiles of more than 1,600 cells from the visual cortex were reported (Tasic et al., 2016). These studies provided information on the transcriptomic diversity of neurons and non-neuronal cell types in the brain and highlighted the power of scRNA-Seq for exploring the CNS. Since these initial studies, both embryonic and adult mouse brains have been extensively studied using single-cell RNA transcriptomic methods, followed by human transcriptomic atlases (Eze et al., 2021). A review article published in May 2023, in Nature Reviews Neurology, highlights all the major findings in the field made possible by scRNA-Seq (and spatial transcriptomics), and it is a wonderful resource for diving deeper into the topic of scRNA-Seq (and its future adaptations) for advancing the filed of neurobiology, so be sure to check it out.
Summary
scRNA-Seq has revolutionized the field of transcriptomics, making it possible to study cellular complexity and heterogeneity, to understand transitional states and all the nuances of gene expression in complex cell populations, tissues and organs. Many fields of biology have flourished as a result of the advances made possible by scRNA-Seq, and in this blog post we focused on discoveries in the fields of immunology, cancer research, development, neurobiology, and diabetes.
As valuable as this next-generation sequencing technology is, it is important to emphasize that there are many different protocols for scRNA-Seq, and they differ not only in the details of the technology and methodology they are based on, but also in their “output” or the depth of detail they can resolve from the mRNA data they generate. It is therefore important to know which method to use for which purpose – and we will cover this topic in our next blog article in the series of blog articles on scRNA-Seq.
References:
2018562 (2018). The Tabula Muris Consortium. Nature 562, 367-372. DOI: 10.1038/s41586-018-0590-4.
Written by Masa Ivin, PhD






