A typical RNA sequencing experiment assesses steady-state levels of the transcriptome by quantifying the total RNA content. This delivers a “snapshot” of the transcriptomic state of a cell at a given timepoint but does not cover the dynamics of the transcriptome. Further, the approach is insensitive towards genes with low transcription rates and genes expressing stable RNAs, and immediate reactions towards internal or external stimuli cannot be resolved.
These limitations can be overcome by methods that utilize the incorporation of labeled nucleotides like 4-thiouridine (S4U) into newly transcribed RNA. The first series of workflows had to rely on biotinylation and pull-down steps to isolate nascent RNA from “old” RNA resulting in high input requirements and long protocols. Elegant, recent developments now combine metabolic RNA labeling with biochemical nucleoside conversion.¹ Thereby, total RNA levels can be quantified in RNA-Seq experiments together with newly transcribed RNA, simplifying the workflow and making the overall process more efficient.
SLAMseq for transcriptome-wide RNA dynamics measurements
The SLAMseq method (thiol(SH )-Linked Alkylation for the Metabolic Sequencing of RNA) is at the forefront of metabolic RNA sequencing by connecting a simple protocol that adds only two steps to a standard RNA-Seq setup, with high efficiency and thorough validation. SLAMseq was developed by the Ameres lab (IMBA – Institute of Molecular Biotechnology, Vienna , Austria) and is available from Lexogen (Vienna, Austria) in kit format and as service. ²
SLAMseq uniquely employs iodoacetamide (IAA) to efficiently alkylate incorporated S4U, converting it into a photo- and heat-stable cytosine analog. The reverse-transcriptase-induced T > C conversion is then detected and quantified by RNA sequencing (Fig. 1).
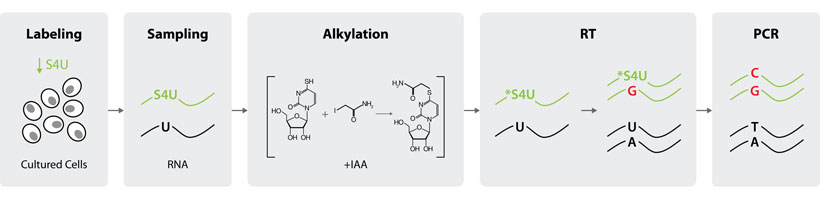 Figure 1 | SLAMseq workflow. Only two steps – labeling and alkylation – are added to a typical RNA-Seq experiment to tag newly transcribed RNA with T>C conversions.
Figure 1 | SLAMseq workflow. Only two steps – labeling and alkylation – are added to a typical RNA-Seq experiment to tag newly transcribed RNA with T>C conversions.
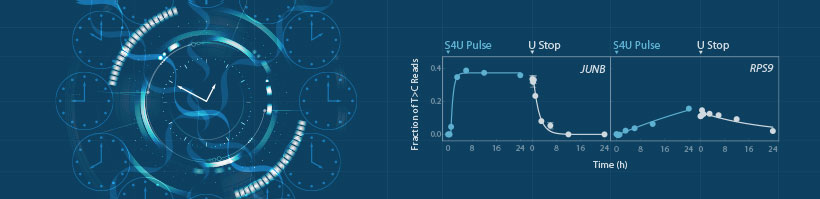
In their first publication describing the SLAMseq method, Herzog et al.,2 sampled RNA at different time points and assessed the fraction of newly transcribed RNA in relation to the total RNA, revealing transcriptome-wide RNA synthesis and degradation kinetics. In doing so, global and transcript-specific transcription rates and mRNA half-lives for mouse embryonic stem cells were derived. The Authors also provided quantitative and mechanistic evidence for transcript-specific RNA turnover mediated by post-transcriptional gene regulatory pathways initiated by microRNAs and N6-methyladenosine.
To obtain the read depth required for detecting T>C conversions also at early time-points and for low expressed genes the study combined SLAMseq metabolic labeling with QuantSeq 3’ mRNA-Seq library preparation (Lexogen, Vienna, Austria). To accurately quantify gene expression, this 3’ tag-profiling method needs only about 10% of the reads typically required by standard RNA-Seq methods covering the whole transcript. Assessing seven time-points within a 24h time period in duplicate was therefore possible in a cost-effective way.
SLAMseq for identifying direct transcriptional targets
In a different application, Muhar and colleagues from the Zuber (IMP – Research Institute of Molecular Pathology, Vienna, Austria) and Ameres (IMBA, Vienna, Austria) labs combined rapid chemical-genetic perturbation with SLAMseq to assess immediate gene expression changes and to preclude indirect effects. 3 In leukemia cell lines, degradation or inhibition of BRD4 revealed global downregulation of transcription while loss of MYC led to a selective decrease in transcription of genes involved in protein and nucleotide synthesis. The combination of SLAMseq with small-molecule-mediated inhibition or degradation provided an effective approach to measure global and specific transcriptional responses to cellular perturbations and to identify primary and secondary transcriptional targets. In this study as well the QuantSeq 3’ mRNA-Seq kit provided the required read depth for gene expression evaluation at minimal costs (see also this blog entry).
SLAM-ITseq: Sequencing cell type-specific transcriptomes without cell sorting
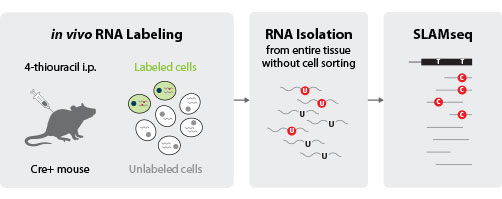
Figure 2 | SLAM-ITseq workflow. Image modified from Matsushima et al., 2018
In a recent publication Matsushima and colleagues now show that metabolic labeling can also be used to accurately capture the in vivo transcriptome of a specific cell type. without prior cellular or molecular sorting. They named the resulting method SLAM-ITseq (IT, in tissue).4 For this approach, 4-thiouracil (note, this is different from 4-thiouridine in SLAMseq) was injected into a transgenic mouse expressing functional phosphoribosyltransferase (UPRT) exclusively in one specific cell type. UPRT enables 4-thiouracil to be incorporated into RNA, by converting it into 4-thio-UMP. Thus, only RNA transcribed in cells that express UPRT gets labeled. The combined SLAMseq and QuantSeq workflow then provides for an easy detection of tissue-specific gene expression and successfully identified the cell type-specific transcriptome in three different tissues: endothelial cells in brain, epithelial cells in intestine and adipocytes in white adipose tissue.
in vivo SLAMseq for small RNA
Tissue-specific SLAM-ITseq can also be used to study small RNA transcriptional regulation. In their paper published in Developmental Cell, Sharma and colleagues used SLAM-ITseq to label small RNAs synthesized in the caput epididymis.5 Tissue-specific labeling was achieved by injecting 4-Thiouracil (4-TU) into Defb41:Cre mice that expressed the UPRT enzyme specifically in the caput epididymis. The authors then performed SLAMseq and small RNA sequencing, on RNA isolated from the testes and caput epididymis of 4-TU-injected and control non-injected mice. Using SLAMseq and small RNA next generation sequencing, labeled microRNAs with U>C conversions were significantly enriched in the caput epididymis of 4-TU-injected mice. This finding provided strong support for the hypothesis that a proportion of miRNAs transcribed in the epididymis are transported via epididymosomes to the sperm as they mature within the epididymis.
SLAMseq kits
Lexogen offers the SLAMseq Explorer Kit to evaluate the best S4U concentration for a not yet assessed cell line and to obtain S4U incorporation rates. The SLAMseq Kinetic Kits then provide the Anabolic Module for measuring RNA synthesis, and the Catabolic Module to measure RNA degradation. A dedicated SLAMseq data evaluation pipeline called SLAMdunk was programmed to calculate background-subtracted, U content- and coverage-normalized T > C conversion rates for each transcript, yielding total and nascent expression values for each gene from one RNA-Seq data set. SLAMdunk pipeline is available at GitHub.
SLAMseq service
Lexogen also offers SLAMseq and SLAM-ITseq as a service with QuantSeq 3’ mRNA-Seq libraries prepared from alkylated RNA provided by the customer. The libraries are sequenced in-house, and NGS data is analyzed using the SLAMdunk pipeline, yielding total gene expression values together with the fraction of transcripts that were S4U-labeled. RNA synthesis and decay rates as well as cell- and tissue-specific expression patterns can be derived from this data.
SLAMseq intro and webinar
Learn more about the development, the technical aspects and the RNA turnover application of SLAMseq in an introductory video from IMBA and in a webinar given by Dr. Stefan Ameres, one of the inventors of SLAMseq.
References
- Baptista, M. A. P. & Dölken, L. (2018) RNA dynamics revealed by metabolic RNA labeling and biochemical nucleoside conversions. Nature Methods 15, 171–172. DOI: 10.1038/nmeth.4608.
- Herzog, V. A. et al. (2017) Thiol-linked alkylation of RNA to assess expression dynamics. Nature Methods. DOI: 10.1038/nmeth.4435.
- Muhar, M. et al. (2018) SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science (New York, N.Y.). DOI: 10.1126/science.aao2793.
- Matsushima, W. et al. (2018) SLAM-ITseq: Sequencing cell type-specific transcriptomes without cell sorting. Development (Cambridge, England). DOI: 10.1242/dev.164640.
- Sharma, U. et al. (2018) Small RNAs are trafficked from the epididymis to developing mammalian sperm. Developmental Cell. DOI: 10.1016/j.devcel.2018.06.023.
Blog article written by Stephanie Bannister and Lukas Paul.






