As the experts in RNA-Seq, we get this question very frequently. In fact, it is a tricky question to answer with confidence. One of the reasons is that RNA-Seq consists of diverse RNA sequencing types and we cannot have one single generalized answer for them.
Two major types of RNA-Seq
What we call an RNA-Seq comprises a huge variety of RNA sequencing projects with different objectives, applications and target RNA (e.g., mRNA, ribo-depleted total RNA, small RNA and so on). However, the most common two types of RNA-Seq may be 1) Whole Transcriptome Sequencing and 2) Expression Profiling Sequencing.
Whole Transcriptome Sequencing aims to sequence entire transcripts in the sample to acquire the most comprehensive level of information for differential expressions, alternative splicing detection and even for the purpose of de novo assembly of the transcripts.
Expression Profiling Sequencing, on the other hand, has as its main objective seeing how genes are expressed over a broad range of samples to identify which genes are up/down-regulated in specific environments, tissues, and cells. This kind of approach used to be done by microarray or quantitative PCR in the past, but NGS is being substituted rapidly as the cost for sequencing has dropped significantly.
Normally, the sequence data from Whole Transcriptome Sequencing comprises the data for expression profiling as well, and that is why it is the most common form of RNA-Seq projects at the moment. If you simply order an RNA-Seq library prep kit without specifying your objective, you will most likely be getting the one for Whole Transcriptome Sequencing.
However, if you are thinking of doing the former and having the latter at the same time, you will have to think again because you will be losing something by doing so. Let’s see what the loss is.
Whole Transcriptome and Expression Profiling – more is not always better.
Below you will find a table which shows how many reads are normally required for different sequencing projects. It is based on the publications which have general consensus among researchers. Nevertheless, it may still not be a golden rule for every sequencing project. What I want to draw your attention to, however, is the relative difference between the Whole Transcriptome Sequencing and Expression Profiling Sequencing.
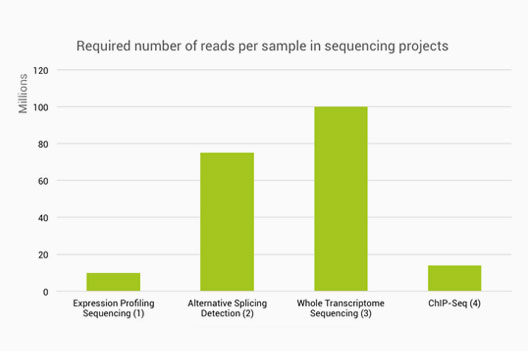
1) Liu Y., et al., RNA-seq differential expression studies: more sequence or more replication? Bioinformatics 30(3):301-304 (2014) 2) Liu Y., et al., Evaluating the impact of sequencing depth on transcriptome profiling in human adipose. Plos One 8(6):e66883 (2013) 3) Bentley, D. R. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59 (2008) 4) Rozowsky, J.et al., PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nature Biotech. 27, 65-75 (2009).
As you can see from the table above, the Whole Transcriptome Sequencing requires about 10 times more sequence reads than the Expression Profiling Sequencing. It means that 90% of your sequence reads are simply redundant or wasted if you choose Whole Transcriptome Sequencing libraries while all you need is just gene Expression Profiling. Why is this happening?
Only one sequence read per transcript. No more is needed for expression profiling.
Normal RNA-Seq libraries are designed to randomly sequence any part of a transcript which may not always be representing the identity of each transcript, meaning that multiple locus in a transcript should be sequenced to identify a transcript. However, if you can read exactly the same locus of many transcripts, just one single read per transcript is enough for the identification. This is what makes for such huge saving of sequence reads in in Expression Profiling sequencing. Simply by choosing expression profiling libraries instead of the ones for whole transcriptome, you can save about 10X of sequencing capacity which can, instead, contribute to higher multiplexing capability which allows more samples to be compared in parallel. For this you need a dedicated library prep which is designed to sequence only the representative position of each transcript. When mapped to the reference and successfully identified, such expression profiling library will allow you to sequence 10 times more samples than the whole transcriptome library.
3’ end of mRNA as an identifier for the transcripts.
We saw that the 3′ end of each transcript can be a good target for this representation. Therefore, we developed a protocol which starts reverse transcription from the poly (A) tail of each transcript followed by random priming for the second strand synthesis. By this simple protocol we could generate libraries which specifically target and amplify the 3’ end of each transcript and are ready to be analyzed on major next-generation sequencing platforms. Most importantly, only one fragment is generated from a transcript, enabling simple counting of NGS reads for fast evaluation of gene expression values.
The advantages are numerous. Such approach increases multiplexing capability which significantly drops the price per sample, enabling high-throughput screening. As only one fragment is sequenced per transcript, there’s no need for length normalization like FPKM and RPKM which normally creates huge constraint in post-sequencing data analysis. Also, short read with single-end sequencing is enough for the purpose which saves sequencing cost dramatically.
Set your RNA-Seq objective first, choose the proper library, and then decide how many reads you will need for it.
So, before you start the RNA-Seq project, it is very important to define the objective of your project, whether to see the entire transcriptome of your samples including splice variants and de novo assembly of transcripts, or to get the gene expression profiles and perform massive screening of diverse samples. Careful consideration of the right RNA-Seq experiment is extremely useful, because here “more is not always better”.
Lexogen provides 3’ end-sequencing library prep kit of QuantSeq. For details about this library prep, please refer to the product information page or Nature Methods publication.
Related links and documents
http://www.nature.com/nmeth/journal/v11/n12/full/nmeth.f.376.html






